

吗啡和芬太尼的对 μOR 的 β-arrestin 蛋白活性的作用
2023-01-05 来源:MedChemExpress 点击次数:1847阿片类药物是治疗急性和慢性疼痛的最有效药物。分别作为具有代表性的阿片类生物碱和合成阿片类药物,吗啡和芬太尼被用于癌痛治疗、麻醉镇痛、预防性镇痛以及术后多模式镇痛。虽然阿片类药物是有效的止痛药,但它们会导致严重的副作用,如呼吸抑制 (呼吸抑制所导致的死亡引发了广泛传播的 “阿片类药物危机”[1,2],尤其是在北美)、成瘾和便秘,从而限制了它们的临床应用。
根据 2019 年发布的报告,超过 70% 的“阿片类药物危机”死亡是合成阿片类药物 (主要是芬太尼及其衍生物) 过量使用所致。这些副作用限制了阿片类药物在临床的应用。
阿片类药物的功能由四种 G 蛋白偶联受体 (GPCRs) 家族介导,即 μ、κ、δ 和伤害感受肽受体 (NOPR),在这些阿片受体中,μ 型阿片受体 (μOR) 被发现是镇痛和副作用的主要受体。有研究指出,阿片样物质诱导的镇痛作用归因于 μOR 的 Gi 蛋白信号转导 (图 1),然而,其副作用 (呼吸抑制等) 究竟是由哪个信号通路产生,目前存在争议:一种观点认为由 β-arrestin 信号转导引起,另一观点认为与 G 蛋白门控的内向整流钾通道 (GIRK) 的信号转导有关。
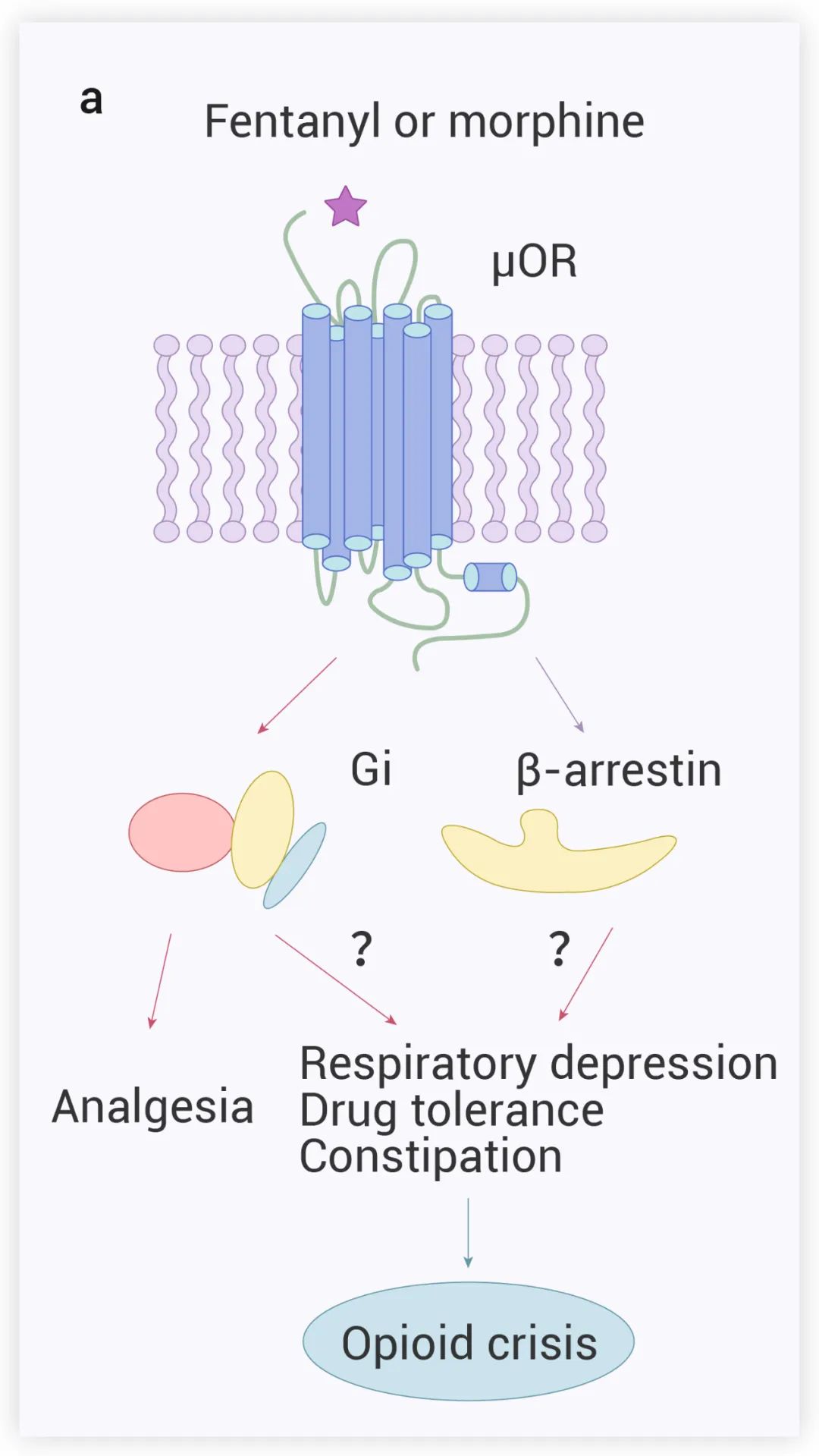
图 1. 芬太尼和吗啡诱导的 μOR 信号传递和潜在的药理作用[1]
如何减少副作用?
今年 11 月,Cell 在线发表了题为 Molecular recognition of morphine and fentanyl by the human μ-opioid receptor 的研究性论文。这篇文章阐述了芬太尼、吗啡和其他 μOR 激动剂与 μOR 的结合方式,并揭示了它们与受体结合的关键差异。该研究还揭示了芬太尼和吗啡对 μOR 的 β-arrestin 蛋白活性起重要作用的结构因素,并为设计有效的、可能更安全的镇痛剂提供了结构模板[1]。
■ 芬太尼和吗啡如何进行信号转导?
该研究首先表征了研究中使用的六种具有不同化学骨架的阿片类药物——芬太尼、吗啡、SR17018、TRV130、PZM21 和 DAMGO (一种拟肽激动剂,可激活 μOR-Gi 复合物,作为对照)——的信号转导情况。
如果所示 (图 2a),所有五个配体都能激活 μOR,以抑制 cAMP 的产生,芬太尼、吗啡和 PZM21 是完全激动剂,而 SR17018 和 TRV130 是部分激动剂。
在 β-arrestin 招募测定中没有检测到 PZM21 的反应信号,而对 SR17018 和 TRV130 只产生微弱的信号。相比之下,吗啡和芬太尼都能诱导强大的 β-arrestin 招募 (图 2b)。总而言之,芬太尼和吗啡不仅能完全激活 μOR,还可诱导强大的 β-arrestin 招募。
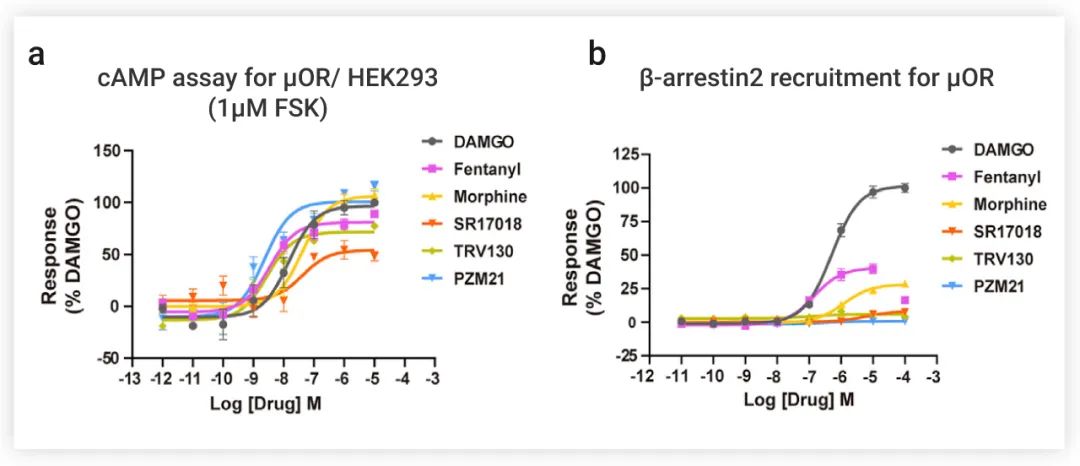
a-b: 阿片类激动剂的 cAMP 积累 (a) 和 β-arrestin2 (b) 招募的剂量依赖反应曲线。DAMGO 为对照物,数据根据 DAMGO 的最大反应进行归一化。
■ 激活 μOR 的关键因素是什么?
芬太尼和吗啡的化学骨架彼此不同,因此,作者分别研究了它们与 μOR 的特异性结合方式。研究表明,芬太尼分子在正位口袋中占据 "Y" 形构象,主要与跨膜结构域 (TMD) 的 TM2、TM3、TM6 和 TM7 的残基接触;吗啡采用椭圆的 "O" 型结构,与来自 TM3、TM6 和 TM7 的疏水残基相互作用 (图 3)。该研究发现,D1493.32 与附近的残基 Q1262.60 和 Y3287.43(DQY 极性模体,以下简称 DQY模体)在两个结构中形成极性相互作用 (图 3a-b),并且 DQY 极性模体的突变,在很大程度上降低了芬太尼和吗啡的 G 蛋白和 β-arrestin 信号转导的效力,这表明 DQY 极性模体对 μOR 的激活至关重要。
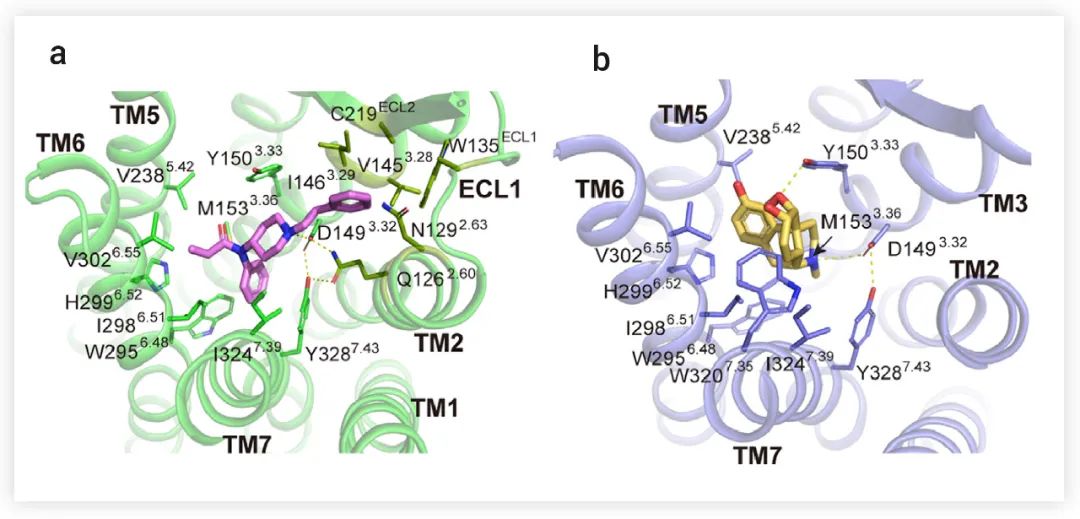 图 3. 芬太尼 (a) 和吗啡 (b) 与 μOR 的相互作用[1]
图 3. 芬太尼 (a) 和吗啡 (b) 与 μOR 的相互作用[1]
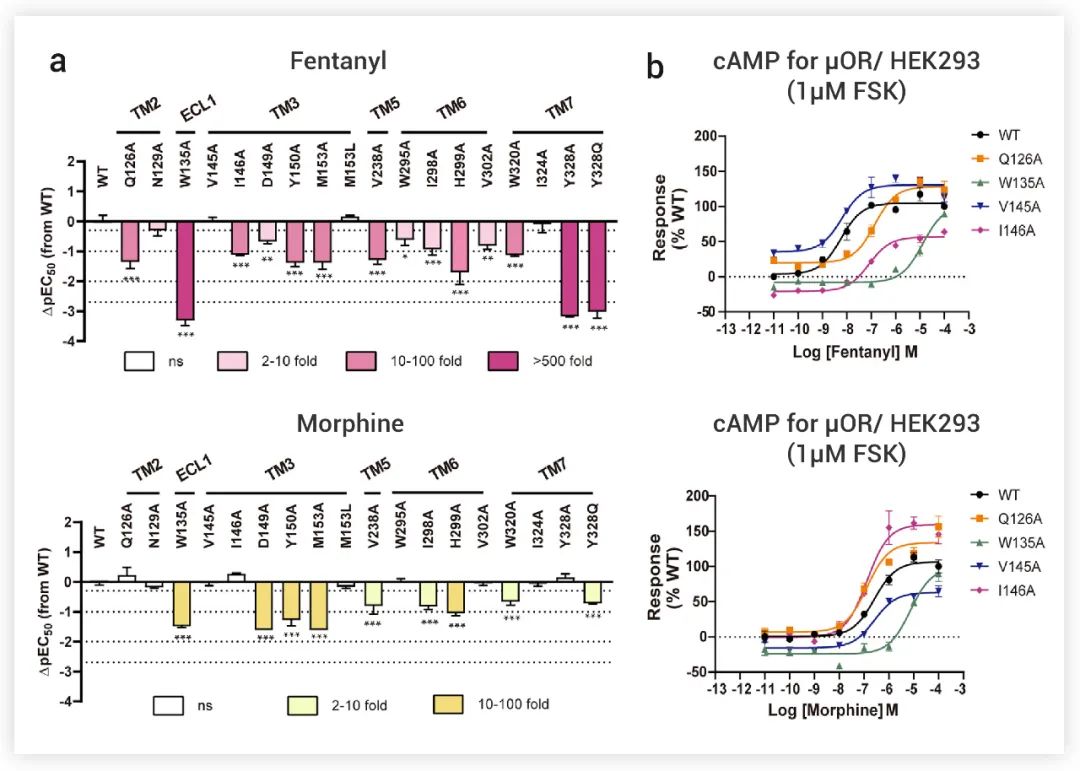
此外,芬太尼和吗啡配体结合袋周围的残基突变,包括 Y1503.33、M1533.36、V2385.42、I2986.51 和 H2996.52,导致两种配体对 G 蛋白和 β-arrestin 信号的活性减弱 (图 4a-b)。
■ 引起 β-arrestin 信号转导的关键因素又是什么?
作者对 μOR 正位结合口袋的残基进行了突变,并测试了这些突变体分别通过芬太尼、吗啡和 DAMGO 激活 G 蛋白信号和 β-arrestin 招募的能力。
结果显示,与 TM2 和 TM3 侧的残基相比,TM6 和 TM7 附近的残基突变对 β-arrestin 信号的影响更为明显。比如,来自 TM6 的 W295A 和来自 TM7 的W320A 突变在 cAMP 积累和 G 蛋白招募方面对 G 蛋白信号转导只有最小或部分影响,而它们几乎取消了芬太尼、吗啡和 DAMGO 诱导的 β-arrestin 招募 (图 5b)。
这说明,配体与 TM6/7 的相互作用对引起 β-arrestin 的信号转导至关重要。
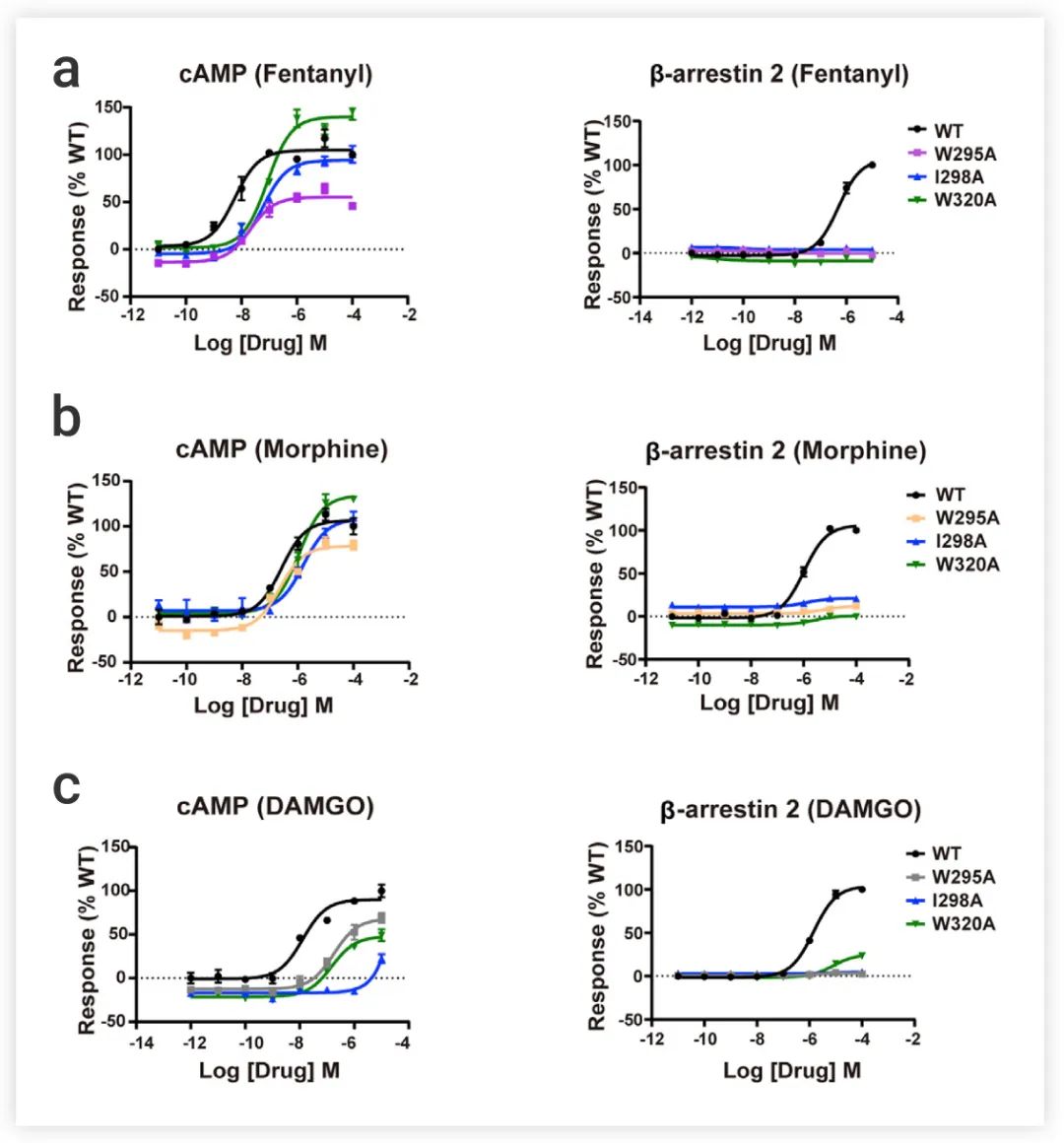
图 5. TM6/7 的代表性残基的突变适度影响了 cAMP 反应,但取消了芬太尼 (a)、吗啡 (b) 和DAMGO (c) 诱导的 β-arrestin 2 的招募[1]
■ 设计分子降低 β-arrestin 活性了吗?为了进一步验证上述发现,作者设计了两个结构相似的芬太尼衍生物 FBD1 (部分激动剂) 和 FBD3 (完全激动剂),以获得 β-arrestin 信号传导减少/消失但 G 蛋白活性相对完整的 μOR 激动剂 (图 6a),也就是说,FBD1 和 FBD3 与μOR 的 TM6/7 之间的相互作用被削弱。
实验结果显示:与芬太尼相比,FBD1 和 FBD3 都显示出 β-arrestin 招募活性的极大降低,并且,FBD3 在 cAMP 抑制或 Gi 招募试验中显示出与芬太尼几乎相同的效力和效率 (图 6b-c)。
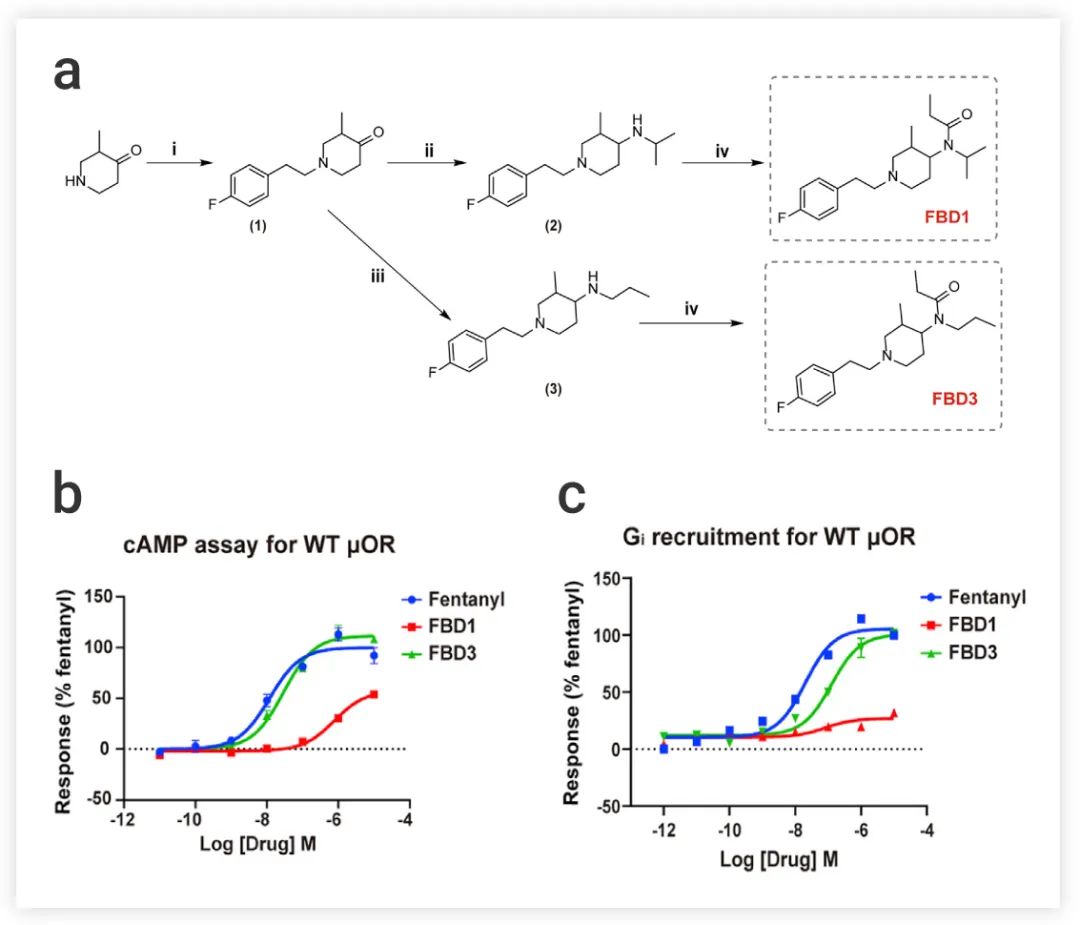
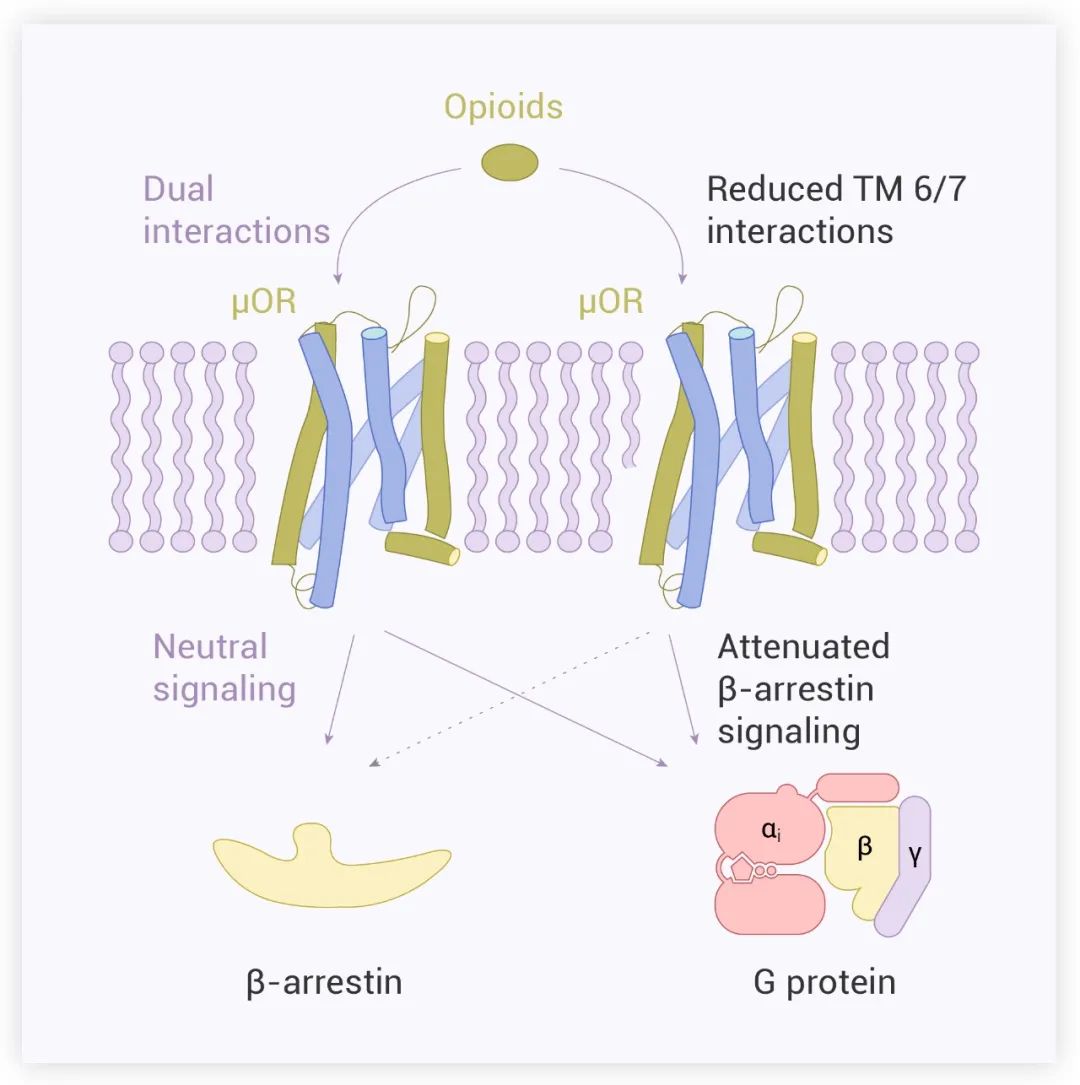 图 7. μOR 的配体诱导的不同信号模型[1]
图 7. μOR 的配体诱导的不同信号模型[1]
这篇文章揭示了人类 μ 型阿片受体对吗啡和芬太尼的分子识别方式,并提出了基于芬太尼结构的类似物设计,削弱 μOR 的 β-arrestin 活性的思路。该研究有助于合理设计下一代镇痛剂,并有望在减少阿片类药物副作用的同时不影响其镇痛作用,甚至增强镇痛效果。
|
相关产品 |
| DAMGO TFA 选择性 μ 型阿片受体激动剂 (Kd = 3.46 nM)。 |
|
选择性 μ 型阿片受体激动剂 (EC50 = 1.8 nM) |
|
μ 型阿片受体激动剂,能与 GTPγS 结合 (EC50 = 97 nM)。 |
参考文献
[1] Zhuang Y, H. Eric Xu, Xin Xie, Ming-Wei Wan, et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell. 2022 Nov 10;185(23):4361-4375.e19.[2] Vadivelu N, et al. The Opioid Crisis: a Comprehensive Overview. Curr Pain Headache Rep. 2018 Feb 23;22(3):16.