

Illumina测序平台在“千人藏族基因组测序”项目研究中的应用
2023-11-13 来源:本站 点击次数:2692
藏族人群对高海拔环境的遗传适应是现代人适应性进化最经典的案例之一。遗传学和考古学研究表明藏族人群祖先早在旧石器时代就迁居到了青藏高原,世代的自然选择使得藏族人群积累了适应性的遗传变异,并获得了生理上的适应特征,包括较低的血红蛋白水平、更强的低氧通气反应、更好的心肺功能和更好的生殖适合度等。
为了能更深入的探究这种适应性遗传变异的秘密,来自中科院昆明动物所和西藏大学的研究人员在国家自然科学基金基础科学中心项目、中科院战略先导专项、中科院青促会项目、西藏自治区重点研发计划和云南省自然科学基金项目的资助下,使用来自Illumina的测序平台进行了“千人藏族基因组测序”项目。该项目发表在Genome Biology(IF:12.3)上。
论文的网址链接为:
https://doi.org/10.1186/s13059-023-02912-1
文章简介
进入基因组时代以来,国内外学者利用全基因组信号扫描找到了一系列在藏族人群基因组中受到正选择的变异位点,共报道了682个基因。然而,由于样本量小、变异位点密度低、检测方法单一等局限,只有EPAS1和EGNL1两个基因能在多项独立研究中成功重复,而其他基因的选择信号仍不确定。
此外,目前的功能研究发现这两个基因只能解释少数几个适应性状,而其他藏族人群高原适应性特征,比如更好的心肺功能,仍无法被目前的正选择基因所解释。因此,在藏族人群高原适应遗传研究的拼图中,目前的数据仍有许多不足和未确定的部分,需要大规模全基因组测序(WGS)数据和综合性达尔文正选择检测方法重新评估和确定。
为了全面评估藏族人群高原适应的遗传位点,中国科学院昆明动物所宿兵团队与西藏大学、西藏阜康医院等单位合作,利用Illumina NovaSeqTM产生了1,001个藏族人群的WGS数据,样本覆盖了中国青藏高原的主要人口分布区。
通过对“千人藏族基因组”数据的系统分析,发现了3500万个变异,其中超过三分之一是新发现的变异。利用大规模WGS数据,基于藏族人群的变异位点频谱和连锁不平衡谱,研究人员构建了首个藏族人群基因组参考面板(1,000 Tibetan-Genome Panel,1KTGP)。
此外,通过使用多信号综合分析(Composite of Multiple Signals,CMS)方法,重新确定了藏族人群基因组中受到达尔文正选择的遗传位点和基因,包括4320个高可信的受选择位点,涉及192个基因。功能注释和富集分析表明,这192个基因涉及多个基因器官和生理系统,支持藏族人群高原适应是多基因效应的假说。研究团队发现了4个具有强选择信号的新基因:TMEM132C、ATP13A3、SANBR和KHDRBS2,与藏族更好的心肺功能相关。该研究基于大规模的藏族全基因组测序数据和发现的适应性基因将成为未来高原地区人群遗传和医学研究的宝贵资源。

研究背景
在最近的人类进化研究中,藏族人对高海拔环境的遗传适应被视为一个经典案例。遗传和考古数据都支持藏族人在高海拔地区的旧石器时代聚居地(超过1000代人)就已经出现雏形,这使得自然选择能够丰富赋予藏族人适应能力的基因突变。这些已知的适应特征包括相对较低的血红蛋白水平,更有效的呼吸,更好的心肺功能,以及更好的生殖能力。
然而,在过去的十年里,尽管科学家们已经进行了许多使用全基因组数据的研究,以寻找在藏族人中显示达尔文阳性选择信号的变异和基因——总共已经报道了682个基因;并且只有两个基因(EPAS1和EGNL1) 在多次研究中被成功复制,其他基因的选择信号仍无法得到验证。此外,已发表的EPAS1和EGLN1的遗传关联分析和功能实验数据似乎只占适应性特征的一小部分,包括保护藏族人免受高原红细胞增多症影响的相对较低的血红蛋白水平和更好的通风。因此,目前的研究仍无法解答藏族的基因适应。
藏族基因适应未解之谜源于当前数据的三大局限性:
(1)样本量小,即所有已报道的藏族全基因组测序(WGS)数据的个体数都不到50个,导致对等位基因频率的估计不准确,检测基因组中选择信号的能力有限;
(2)变异密度低,即已报道的大样本研究只产生覆盖有限基因组的SNP阵列数据;
(3)在检测自然选择信号方面存在偏差, 即已发表的研究只采用一种或两种方法(但研究不同)来寻找选择信号,研究结果不一致。
方法
取样及测序
在中国西藏自治区拉萨某医院(海拔3650 m)共招募1064名受试者。这些被招募的藏族女性均无亲属关系(来自医院妇产科),她们来自83个不同的地理位置(海拔范围:2300-4900米)。每位受试者均获得书面知情同意。本研究方案经中国科学院昆明动物研究所内审委员会审核通过(批准号:SMKX-20160311-45)。采集血样,通过prefill Blood DNA Kit-DUO (KFRPD801212)提取基因组DNA,在Illumina NovaSeq™平台上进行WGS,平均每人40 Gb (11.8×depth)数据(图1B)。在测序和分析之前,为了保护隐私,所有样本都被剥离了个人标识符。所有程序都符合人体实验负责委员会的道德标准。
结果
1,001名藏族人的全基因组测序
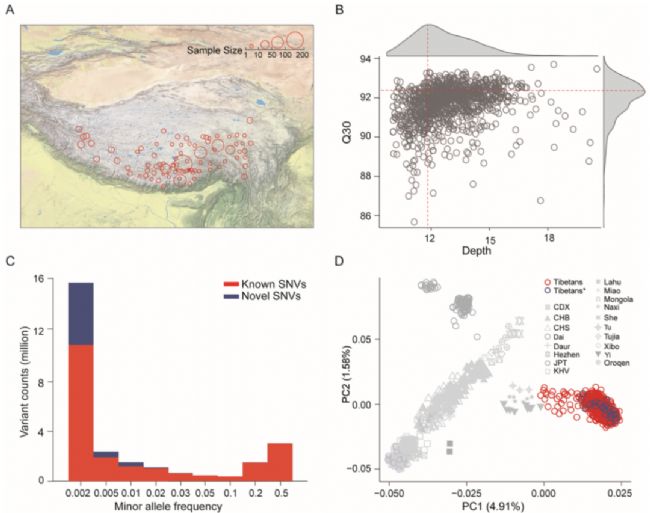
图1:抽样调查藏族地理位置及WGS数据质量评估
A 本研究中藏族样本的地理位置。标明了抽样地点和样本量。
科研人员招募了来自青藏高原83个不同地理位置(海拔范围:2300-4900米)的1064名藏族参与者。
B 藏区1001WGS数据的质量,反映在深度和Q30值上。平均深度和Q30用红色虚线表示。
共计1064名个体的基因在Illumina NovaSeq™平台上进行了WGS测序,测序平均深度为11.8×,数据质量平均Q30为93%。经过严格的过滤,最终保留了1001个个体的WGS数据,用于全基因组变异调用和下游分析。
C 所有已鉴定SNV的次要等位基因频谱。已知的和新的变种分别以红色和蓝色显示。
使用标准的GATK流程,鉴定出3470万个变异,其中包括2990万个单核苷酸变异(SNV)和480万个INDELs(插入和缺失大小小于50bp)。在2820万个双等位SNV中,有36%是没有在数据库dbSNP (版本154)中报告的新SNV。
D 藏族和18个东亚代表性群体的全基因组主成分分析图。红圈是本次研究的1001个样本(藏族),蓝圈是已发表的33个WGS样本(藏族*)
通过主成分分析(PCA),包括1000基因组计划第三阶段(简称1KGP3)和人类基因组多样性计划(HGDP)的1001名藏族人和6527名全球个体,以及33名已发表的藏族人。当只包括东亚人口时,1001名藏族人与33名藏族人紧密地聚集在一起,他们一起形成了一个独立于其他东亚人口的群体,且来自中国的三个民族(土族、纳西族和彝族)与藏族人关系较近。这三个民族生活在青藏高原周围与藏族人杂居。藏族的全基因组杂合率为1.41±0.046,藏族与汉族(藏汉)的遗传距离为0.0095。
藏族人的变异频率和连锁不平衡谱系
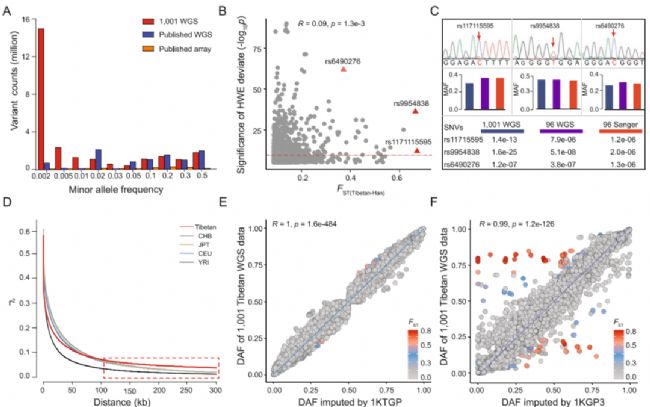
图2:藏族的全基因组变异频率和LD谱系
A 1001 WGS数据和公共的数据之间的MAF的SNV计数比较。1001个WGS数据在检测罕见变异方面比公共的数据要强大得多。
基于1001个基因组序列建立了西藏人的全基因组变异频率和LD谱系与以往小样本量的西藏WGS研究或大样本量的阵列数据相比,该的变异集在检测罕见变异(小等位基因频率,MAF<3%)和全基因组的无偏覆盖方面表现出显著的能力
B 人群间差异较大的SNVs(FST(Tibetan-Han))的HWE偏差分布,HWE偏差的卡值为1e-6。
对藏族人的全基因组变异进行了Hardy-Weinberg平衡(Hardy–Weinberg equilibrium ,HWE)偏离测试,有207个变异显示出明显的HWE偏差(P<1e-6)。同时,它们也显示出藏族和汉族之间的深度分化,这是强烈自然选择的表现(FST(Tibetan-Han)>0.2)。这些变异的质量都很稳定,并通过了所有严格的质量控制,包括读深度、映射质量(MQ)、碱基质量(BQ)和基因型质量(GQ)。
C 通过Sanger测序验证三个具有高FST(Tibetan-Han)的HWE偏移的SNVs(藏族-汉族)。中间的直方图表示三个数据集中三个SNVs的小等位基因频率(MAF),包括1001个藏族人的WGS数据(蓝色),1001个WGS数据中的96个随机样本(绿色),以及96个样本的Sanger测序数据(红色)。直方图下的p值表示基于三个数据集的三个SNPs的HWE偏差的显著性水平。
为了进一步验证这些HWE偏移的变异,并排除测序错误的可能性,研究人员选择了三个具有高FST(Tibetan-Han)值的变异(rs117115595,rs9954838和rs6490276),并在在96个随机选择的藏族人中进行Sanger测序。三个被选择的变异的基因型频率和HWE模式与WGS数据高度一致,表明HWE偏移的变异很可能是在藏族中经历了正向选择的真正突变,而不是基因分型错误。结果表明:对于经历强烈自然选择的人群(如西藏人),HWE偏差的显著性阈值应该更宽松。
D 藏族和世界其他人群的LD衰减模式的比较。虚线框表示藏族人独特的LD衰变模式。对于长基因组区域(>100kb)的衰变,藏族显示出比世界其他人群更慢的衰变(反映在更高的r2值),这是扩展单倍型同质性的表现。
E 来自1001个西藏WGS数据和3008个西藏阵列数据的全基因组SNVs的派生等位基因频率(derived allele frequency, DAF)的相关性,由1KTGP进行归因。
利用获取的1001个基因组序列重建了一个西藏特有的LD参考(简称1KTGP),并对已发表的使用全球人群参考的阵列数据(简称1KGP3)的归因准确性进行了评估。结果发现,1KTGP估算的基因型频率与1,001 WGS数据高度一致。
F 用1KGP3归因时的相关图。
1KGP3计算的基因型频率有许多SNVs与1001 WGS数据有严重偏差,例如位于EPAS1基因区域的SNVs。
重新定义藏族人的正向选择的基因组特征
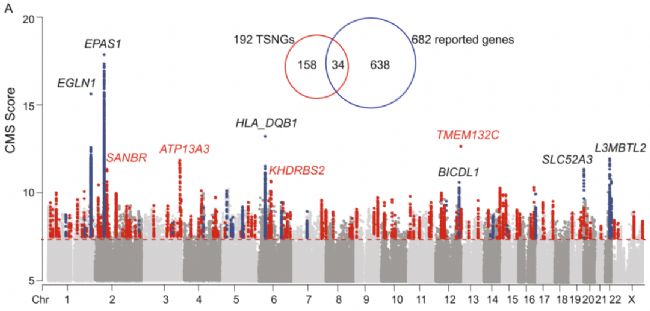
A藏族全基因组SNVs的CMS评分分布。192个前导基因区域(lead gene regions)用红色(新发现的基因)和蓝点(已报道的基因)分别标记。排名前10位的TSNGs以基因名称标示(4个新发现,6个已报道)。维恩图显示了本研究中报告的基因集和鉴定的基因集之间的重叠。
科研人员通过多种信号复合(Composite of Multiple Signals ,CMS)的方法来识别正选择下的变异体。首先生成了具有最高1‰CMS得分(>7.66)的变异集,然后对该变异集进行过滤,只保留显示西藏特有富集的变异,即与全球四个主要人群(汉族、日本人、欧洲人和非洲人)相比,西藏人的富集等位基因频率更高。最终的数据集包含了4320个被认为是西藏选择主导的SNVs(Tibetan selection-nominated SNVs,TSNSs)变异。它们位于236个独立的基因组区域,由192个主导基因代表,被称为西藏选择指定基因(TSNGs)。在这192个TSNGs中,34个是以前报道过的,其他158个是新发现的基因。
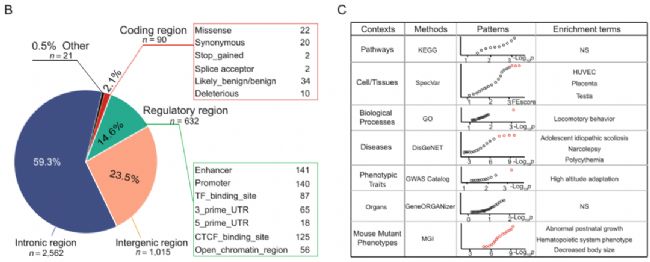
B 4320个TSNGs的功能注释。“调控区”是指带有调控注释的非编码区。
通过对4320个TSNSs进行功能注释发现,大部分(75.4%)位于非编码区,632个(14.6%)位于被注释的调节区。编码区有90个TSNSs,包括22 missenses(错义突变), 20 synonymous(同义突变), 2 stop-gained(外显子提前终止翻译), 2 splice acceptor(剪接受体), 10 deleterious(有害突变), and 34 likely_benig/benign(可能是良性突变)。
C不同方法下TSNGs的功能富集模式。在气泡图中,有意义的项用红色表示。
使用7种方法进行功能富集分析发现:在细胞类型和组织方面,这些TSNGs在人脐静脉内皮细胞(HUVECs)、胎盘和睾丸中显著富集,反映了藏族人在氧感(内皮细胞)、发育(胎盘)和生殖能力(睾丸)方面的适应性变化,与小鼠敲除表型(出生后生长异常和体型减小)以及疾病期(青少年特发性脊柱侧凸)一致。新发现的322 bp内含子缺失PKHD1L1 (PKHD1 ciliary IPT domain containing fibrocystin/polyductin like 1;含 PKHD1 纤毛 IPT 结构域纤维囊蛋白/多管蛋白)在藏族人群中富集(49.5%),但在非藏族人群中相对较少(<23%)嗜睡症通路富集结果相呼应。
表1:藏族人中14种高丰富度的错义变体
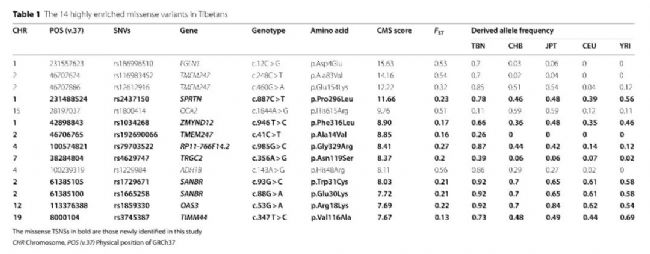
粗体显示的错义TSNGs是本研究中新发现的
在22个错义TSNS中,有13个在藏族中高度富集,与全球其他人群相比,在藏族中的频率>20%,其中4个是以前报道过的,包括EGLN1(rs186996510)、TMEM247(rs116983452和rs12612916)、ADH1B(rs1229984)和OCA2(rs1800414),而其他10个是本研究中新发现的。例如,rs79703522是RP11-766F14.2(编码一种在肌肉、心脏和肾脏中表达的功能未知的蛋白)的错义突变,在藏族人中占优势(87%),平均比全球其他人群高59%。值得注意的是,在之前的研究中,有54个频率丰富的错义和功能缺失突变被报道,这其中只有5个可以在本次的大规模WGS数据中成功验证(FST(Tibetan-Han)>0.1),这表明大样本量对于准确估计等位基因频率至关重要。
新发现的顶级TSNSs解释了西藏人心肺功能的适应
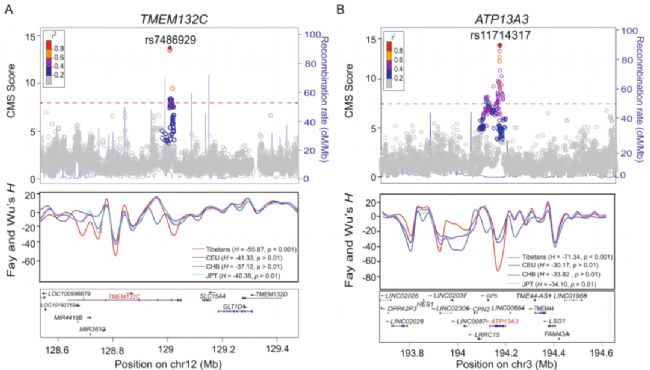
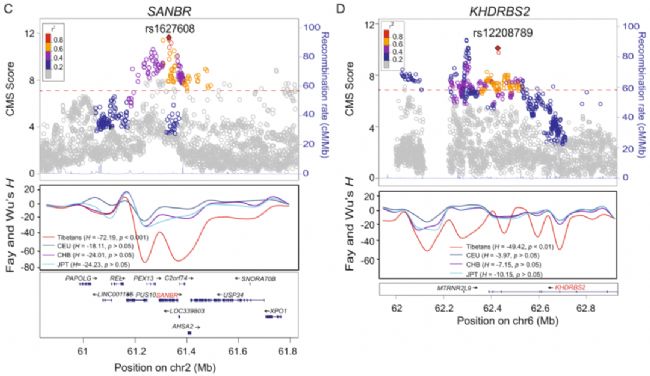
A-D CMS评分和重组率的区域图,其中的峰表示选择信号。峰值SNVs用颜色标记。并给出了四种基因的滑动窗Fay和Wu’s H检验结果。计算的重组率(r2)表示峰值SNV与其他SNVs之间估计的连锁不平衡(linkage disequilibrium,LD)程度,并以颜色编码。CMS=7.66(前1‰)的显著性阈值用红色虚线表示。H值为给定区域(红色标记)的最大得分,覆盖了4个基因SNVs峰值的上、下游500 kb区域
表2西藏基因组中的十大选择信号
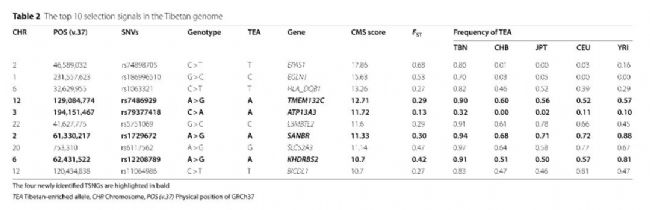
粗体显示的TSNGs是本研究中新发现的4个
在前10个TSNGs中,除了先前报道的6个基因(EPAS1、EGLN1、HLA_DQB1、L3MBTL2、SLC52A3和BICDL1)外,我们还发现了4个具有强选择性的新基因,包括TMEM132C、ATP13A3、SANBR和KHDRBS2。
A TMEM132C基因区
TMEM132C(跨膜蛋白132C)是TMEM132家族的成员,其分子功能尚不清楚。峰SNV rs7486929 (CMS=12.71) 位于TMEM132C的内含子区,具有藏族特异性富集(ΔDAF>29%)。Fay和Wu的H检验进一步证实了TMEM132C的选择信号(H= - 55.87, p<0.001)。之前基于家族的研究中报道了该基因的突变与肺和肺功能(1 s用力呼气量(forced expiratory volume in 1 s ,FEV1))有关。
B ATP13A3基因区
ATP13A3 (ATPase 13A3)是P型ATP酶家族的一员,可跨细胞膜运输多种阳离子,是哺乳动物多胺运输系统的主要组成部分。藏族人与其他人群相比,排在最高的SNV rs11714317表现出强烈的选择特征和独特的LD衰减模式(CMS=11.7, XPEHH=5.7)。Fay和Wu的H检验进一步验证了ATP13A3的选择(H= - 71.34, p<0.001)。ATP13A3在肺血管重构和肺动脉高压(pulmonary arterial hypertension ,PAH)中发挥重要作用。西藏富集的ATP13A3突变可能能保护西藏人免于肺动脉高压(PAH)。
C SANBR基因区
SANBR (CSR的SANT和BTB结构域调节因子,也称为KIAA1841)因其与过氧化物酶体生物发生障碍有关而闻名。SANBR的选择性信号在各种统计数据中是一致的,包括Fay和Wu’H检验(H= - 72.19, p<0.001)。排名最前的SNV rs1627608在藏族和汉族之间存在高度分化(FST(Tibetan-Han)=0.25),根据GTEx数据库,它是睾丸、肺、动脉-主动脉、肌肉-骨骼和心-房附件的表达数量性状位点(expression quantitative trait locus,eQTL)。
D KHDRBS2基因区
KHDRBS2 (KHRNA-binding domain containing, signal transduction associated 2)是一种参与选择性剪接调控的RNA结合蛋白,在肺和脑中大量表达。排名最前的SNV rs12208789位于KHDRBS2的内含子区,在藏族人中表现出强烈的正选择,在藏族人中的频率比全球其他人群高32%。大规模人口GWAS研究显示,KHDRBS2与肺功能(FEV/FEC比值)和房间隔缺损相关。
西藏人遗传适应的多基因和多效性效应
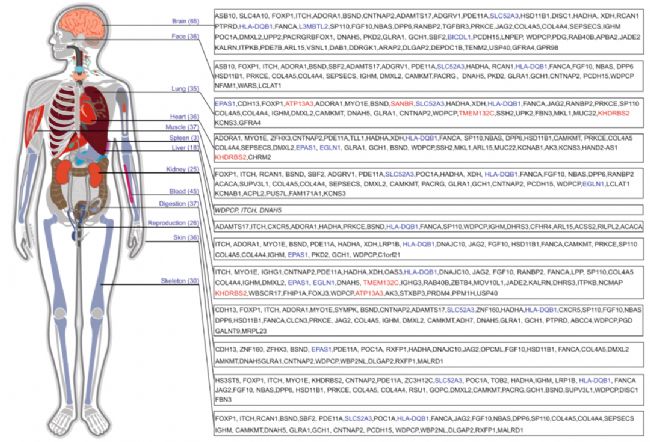
通过GeneORGANizer,将基因在现有功能数据库的基础上分配到不同的器官或生理系统。前10个TSNGs以红色(新发现)和蓝色(已报道)的粗体突出显示。
通过GeneORGANizer分析来确定192个在本研究中已鉴定的TSNGs如何促进西藏人各种生理系统的适应。结果表明,这些TSNGs在多个器官/系统中起作用,包括藏族人已知具有适应性特征的器官/系统,如血液(45个基因)、肺(35个基因)、心脏(36个基因)和生殖(26个基因),以及那些与适应没有已知联系的器官/系统,如大脑(65个基因)、面部(36个基因)、肌肉(37个基因)、肾脏(25个基因)、消化(37个基因)、皮肤(36个基因)和骨骼(30个基因)。值得注意的是,许多TSNGs似乎在多个器官/系统中起作用,例如,HLA-DQB1是排名前十的TSNGs之一,由于其在免疫系统中的作用,它几乎在所有列出的器官/系统中都起作用。同样,SLC52A3出现在7个不同的器官/系统中,这意味着SLC52A3除了在大脑中已知的作用外,它还可能有助于其他器官的适应。
结论
通过这个研究,科研人员利用Illumina NovaSeq™生成了大规模的藏族WGS数据,并为藏族人群提供了特定人群的参考。在这份大规模的藏族WGS数据中,鉴定出了一组具有正选择信号的高置信度基因(192个TSNGs)。这些基因可能在人体的多个器官/系统中发挥多基因和多效性作用,它们可能共同作用形成西藏人的适应性状。这些发现证明了大规模WGS数据在群体研究中的巨大价值和潜力。
相关文章
更多 >