

全基因组测序在血液恶性肿瘤中的应用:基因组时代髓系和淋系肿瘤分析
2024-12-23 来源:Illumina因美纳 点击次数:2619- 研究人员针对急性髓系白血病、骨髓增生异常综合征、骨髓瘤和慢性淋巴细胞白血病等癌症的研究大大改善了个性化治疗。
- 传统的检测方法包括核型、FISH(荧光原位杂交)、染色体微阵列芯片和基因panel。
- 全基因组测序只需一个工作流程就能检测出与这些疾病相关的所有关键异常和变异类型。
简 介
1973年,芝加哥大学的Janet Rowley发现了慢性髓系白血病(CML)的细胞遗传学基础——在她研究的几乎每一个细胞中,都发现了9号染色体和22号染色体之间的易位。这一发现建立在1959年Peter Nowell和David Hungerford对CML患者费城染色体的鉴定之上,这标志着首次发现与癌症的直接遗传联系。20世纪90年代,分子表征发现,这种相互易位事件会产生一个融合基因——BCR-ABL1,它来自B细胞受体BCR和ABL1的激酶结构域。BCR-ABL1融合基因的鉴定为靶向治疗铺平了道路。伊马替尼是首个针对CML中BCR-ABL1蛋白开发的酪氨酸激酶抑制剂。该药物于2001年获得美国食品和药物管理局批准,标志着癌症治疗进入了一个新纪元,在CML患者中取得了前所未见的成功,五年生存率高达89%。
大多数CML病例(超过95%)都具有典型的t(9;22)易位特征。其他髓系恶性肿瘤,如急性髓系白血病(AML)和骨髓增生异常综合征(MDS),以及淋巴系恶性肿瘤,如多发性骨髓瘤(MM)和慢性淋巴细胞白血病(CLL),都具有遗传异质性的特点,变异种类繁多。例如,在AML中,主要的遗传学发现包括反复的染色体易位,如t(8;21)、inv(16)和t(15;17),以及FLT3、NPM1、CEBPA、IDH1/2和DNMT3A等基因的突变,这些发现定义了具有不同预后影响的特定AML亚型。这些基因改变会影响疾病行为、治疗反应和风险分级。这种遗传结果的多样性是其他髓系肿瘤和淋巴细胞白血病的典型特征。
白血病治疗的一般工作流程包括通过血细胞计数和骨髓活检进行初步诊断、分类、使用基因组工具进行风险分级,以及通过化疗、干细胞移植和维持疗法进行治疗。适当的分类和风险分级对患者管理至关重要,对干细胞移植和其他干预措施的关键临床决策也是必不可少的。
目前,血液肿瘤的基因和分子检测采用多模式方法,这些实验室结果被用于根据世界卫生组织和其他临床指南对疾病进行分类和风险分级。传统的细胞遗传学(核型分析)用于检测染色体变异,而荧光原位杂交(FISH)则有助于确定特定的重排和隐匿性易位。聚合酶链反应(PCR)和逆转录PCR可检测特定基因突变(如FLT3、NPM1)和融合转录本,有助于诊断和监测微小残留病(MRD)。新一代测序(包括靶向panel)为风险分级提供了全面的突变分析。染色体阵列和光学基因组图谱方法可在特定情况下使用。
事实证明,全基因组测序(WGS)是一种高效的替代方法,它能提供全面的基因组图谱,检测所有相关的基因异常,包括染色体变化、结构变异以及细胞遗传学和FISH等传统方法可能遗漏的隐性突变。由此实现了更有效的分类和风险分级,一项针对AML/MDS的研究表明,与目前的标准检测方法相比,使用WGS时,近25%的患者获得额外的诊断结果,约17%的患者获得不同的风险分级1。WGS还能帮助新兴领域(如基于血浆样本的MRD检测)摆脱使用骨髓穿刺液进行检测的做法。WGS通过统一的工作流程,将多项检测合并为一项检测,简化了诊断流程,周转时间更短,仅需五天左右。虽然WGS在历史上一直被认为是一种昂贵的检测方法,但测序成本的降低使其变得更加经济可行。此外,该方法已被纳入美国国立综合癌症网络AML/MDS管理指南,并享受医疗保险报销。
血液肿瘤WGS概览
对血液恶性肿瘤进行全面的基因组分析需要评估多种变异类型,包括小变异(单核苷酸变异和插入缺失小于50 bp)、拷贝数改变(CNA)、结构变异(SV)(如易位)和杂合性缺失(LoH)计算。这些海量信息的分析离不开强大的数据分析能力。DRAGEN(Dynamic Read Analysis for GENomics) 是由Illumina因美纳开发的高性能生物信息分析平台,依托全基因组、全外显子组、转录组、甲基化、单细胞等分析模块,支持精准高效的基因组数据分析。
针对血液肿瘤,DRAGEN Somatic WGS Heme Tumor Only方案结合使用变异检出、覆盖深度分析、断裂端检测和其他算法,可为血液恶性肿瘤相关变异提供准确而全面的结果。
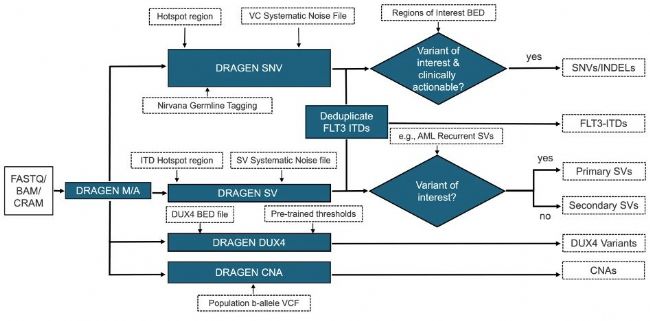
图1:适用于DRAGEN Somatic的DRAGEN Somatic WGS Heme Tumor Only方案
体细胞变异的检测能力在很大程度上取决于样本的测序深度。在图1中,我们对从癌症参考细胞系(Seraseq Myeloid Mutation DNA Mix、NOMO-1、Kasumi-1和HCC1187)中提取的DNA进行了滴定实验,以计算不同覆盖深度下变异的检测限(LoD)。140×的覆盖度可实现等位基因频率大于或等于5%的小变异检测限。为保持一致性,表1进一步详细列出了在140×覆盖度下评估时其他变异类型的LoD。如图2所示,根据特定研究应用的需要,可以通过增加或减少测序覆盖度来改变LoD。
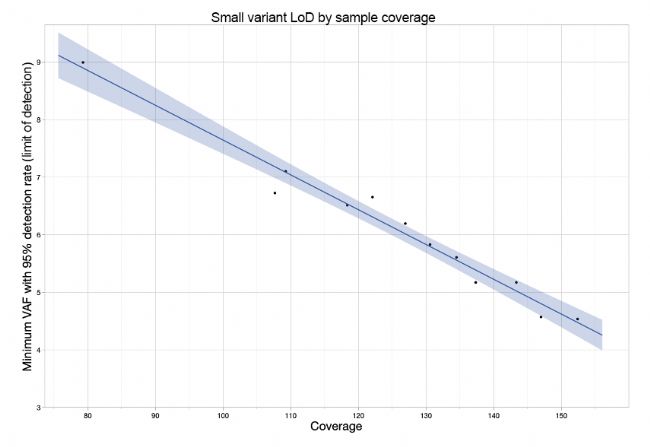
图2:不同覆盖深度小变异的检测限分析灵敏度(检测限)
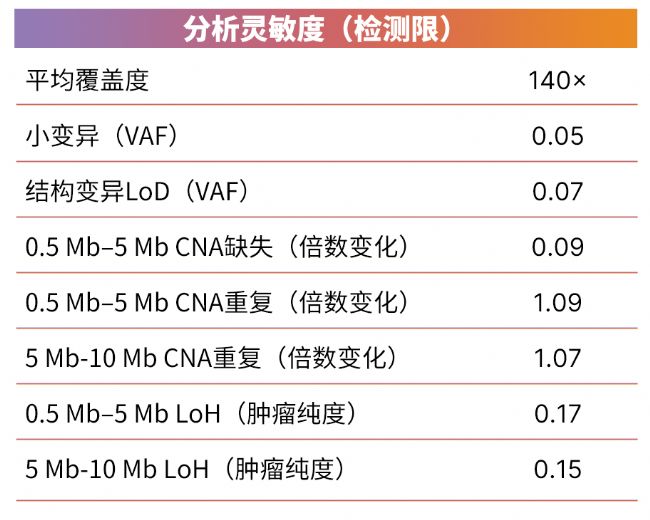
表1:140×覆盖度下确定的小变异、结构变异、CNA和LoH的分析灵敏度(检测限)

我们与研究人员合作,对23个患有血液恶性肿瘤的临床样本进行了PCR-Free WGS,平均覆盖度约为220×(见图3)。我们的团队在这组样本中正确检测出所有临床相关的小变异(n=73,包括4个FLT3-ITD)、结构变异以及超过96%的CNA。具体来说,共评估了两个长度分别约为45 Mb和56 Mb的倒位、8个易位、22个从4.5 Mb到100 Mb的CNA和7个全染色体事件。
为了进一步评估WGS的性能,我们评估了另外30份AML研究参与者的外周血或骨髓穿刺液标本,并将WGS的变异检出结果与肿瘤全景变异分析(“NGS大panel”)TruSight Oncology 500的结果进行了比较。表2汇总了此项研究的结果,并显示了相关变异类型的高性能。

表2:研究队列中约200×覆盖度的WGS检测性能
基因组时代
发表在《血液》(Blood)杂志上的一篇论文指出:“在髓系肿瘤患者中,全基因组测序有可能取代传统的细胞遗传学和测序方法,提供快速、准确的全景变异分析”2。
与传统方法相比,因美纳针对血液恶性肿瘤的全面全基因组测序和信息学解决方案可在单一平台上实现统一的工作流程,从而提高实验室效率和临床疗效。从大型染色体重排到单核苷酸变异,每一个细节都可以通过这一新技术平台进行准确评估。
如何开展?
现有的因美纳产品和工作流程可用于从血液恶性肿瘤患者的标本中生成全面、高质量的基因组。根据研究情况,从外周血和/或骨髓穿刺样本中提取的DNA适合作为起始材料。对于体细胞变异的检测,建议采用更深的平均覆盖度,因为关键事件可能只出现在部分细胞中。为了达到更高的测序深度,通常使用相对较少的文库进行混合。

表3:可优化血液恶性肿瘤WGS研究的现有因美纳产品
参考文献1.Duncavage EJ, Schroeder MC, O’Laughlin M, et al.Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers.N Engl J Med.2021;384(10):924-935. doi:10.1056/NEJMoa2024534
2.Duncavage EJ, Bagg A, Hasserjian RP, et al.Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia.Blood.2022;140(21):2228-2247. doi:10.1182/blood.2022015853
相关文章
更多 >