

Nature Reviews:新方法学(NAMs)在药物非临床安评中的应用
2025-05-27 来源:本站 点击次数:1632原文聚焦四类药物(动物无靶点、跨物种靶点差异、非哺乳动物靶点、临床未预测毒性)的人体体外模型(如器官芯片、类器官)应用案例,强调其在预测人体毒性、补充动物模型局限性方面的价值,并探讨风险评估策略及跨行业联盟对NAMs标准化的推动作用,旨在促进更具人体相关性的安全评估,最终减少药物开发中的动物使用。
新方法学(NAMs)在候选药物非临床安全性评估中的应用正推动监管指南修订,减少动物实验依赖。

一、传统动物模型的局限性与 NAMs 的兴起
- 动物模型的不足:
- 物种差异(如 ADME 参数、基因表达)导致临床前动物研究无法预测人体毒性,例如动物未检测到的肝毒性在人体试验中出现。新型疗法(如细胞治疗、双特异性抗体)因靶点人特异性,动物模型无效(如啮齿类不表达 CEA 抗原)。
- NAMs 的技术突破
- 微生理系统(MPS):如肝芯片、心脏芯片,模拟人体器官微环境,用于评估药物毒性。
- 类器官与 3D 培养:肠类器官联合免疫细胞,预测 T 细胞疗法的肠道毒性。
- 硅基智能工具:如 DILIsym 模型模拟肝毒性机制,BLAST 分析评估靶点同源性。
二、四类药物的体外评估策略与案例
三、案例研究:通过具体的案例研究(如IMCgp100、MEDI-565等药物),展示了如何应用NAMs来评估药物安全性,并在某些情况下减少或避免动物实验。
3.1 IMCgp100
IMCgp100(tebentafusp)是一种融合蛋白,由可溶性、亲和力增强的T细胞受体(TCR)和抗CD3单链抗体(scFv)组成,旨在将表达gp100表位的肿瘤细胞和表达CD3的T细胞结合在一起,用于治疗表达gp100的肿瘤。其药物目标在动物物种中缺乏表达或同源性不足,因此没有相关的动物模型可以用于评估靶向依赖(在靶)毒性。
- 非临床安全性评估方法:Harper等人开发了一套基于商业可用的2D人类癌细胞系的体外评估。用于评估疗效的实验使用抗原特异性、与适应症相关的肿瘤细胞,通过测量乳酸脱氢酶(LDH)释放和凋亡来评估靶细胞杀伤情况。基于干扰素-γ(IFNγ)释放和体外肿瘤细胞杀伤的数据,有助于计算最小预期生物学效应水平(MABEL)。安全性评估中,识别了表达抗原的人类源性黑素细胞作为靶细胞,并使用单一供体的PBMC作为效应细胞,来研究在靶/非肿瘤活性。通过人类全血的细胞因子释放实验(CRA)和血小板激活实验来评估非靶向或非肿瘤效应,以及评估广义人类细胞的风险。
- 研究结果与意义:通过该体外评估预测了IMCgp100的MABEL,并确定了首次人体试验的安全起始剂量,最终该疗法成功进入临床开发。
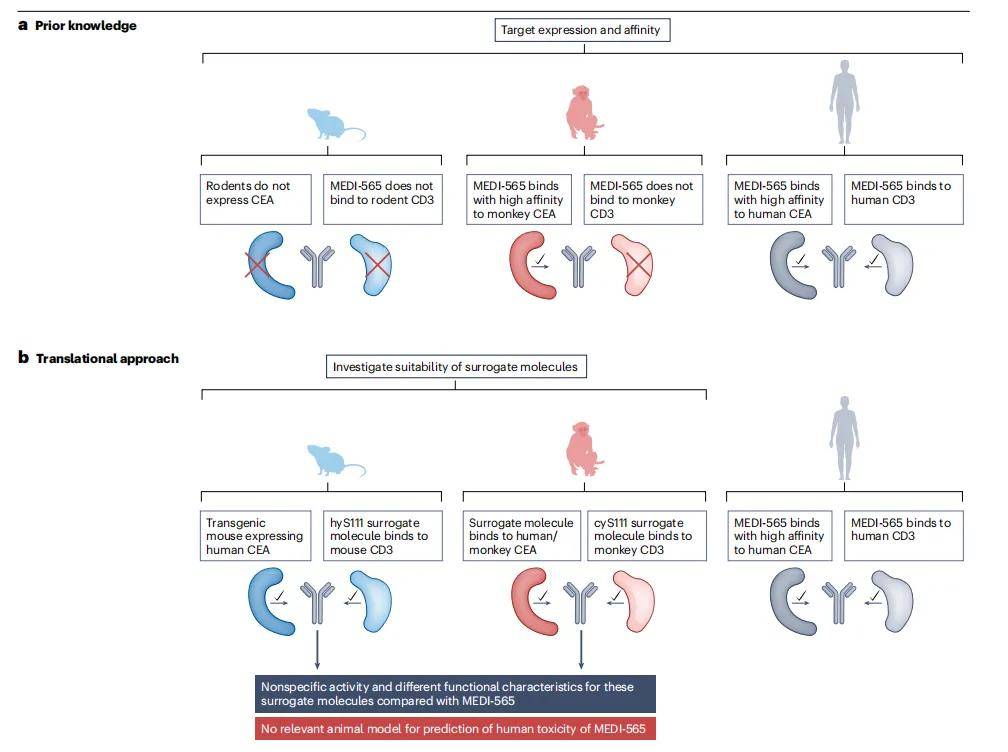
3.2 MEDI-565
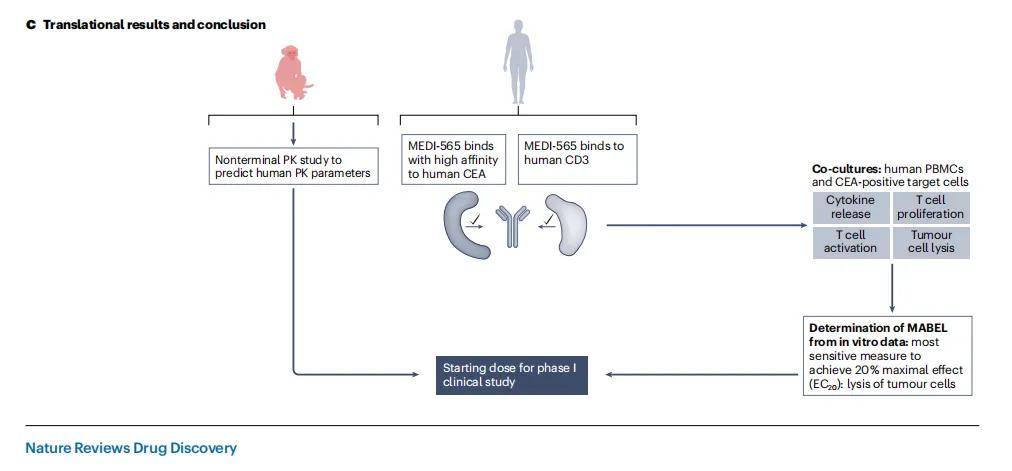
MEDI-565是一种新型双特异性T细胞接合抗体(BiTE),旨在将表达癌胚抗原(CEA)的癌细胞与表达CD3的T细胞结合。由于MEDI-565与人类和食蟹猴CEA结合,但不与食蟹猴CD3结合,且啮齿动物不表达CEA,因此没有相关的动物模型可以用于评估其毒性。
- 非临床安全性评估方法:MEDI-565的非临床安全性评估方案中使用了在食蟹猴和人类CEA转基因小鼠中使用的替代双特异性抗体,但这些替代分子与MEDI-565的特性不同,最终决定不进行任何体内毒理学研究,而是采用仅体外方法,并确定MABEL。通过在共培养中使用人类PBMC和CEA阳性人类肿瘤靶细胞,结合多个人类供体的终点,包括细胞因子释放、肿瘤细胞裂解、T细胞激活和增殖来确定MABEL。
- 研究结果与意义:体外研究结果表明,MEDI-565能够诱导T细胞增殖,证明了细胞因子释放需要同时结合T细胞上的CD3和靶细胞上的CEA,并展示了肿瘤细胞裂解是最敏感的衡量指标,从而确定了MABEL。结合从食蟹猴中获得的非终末药代动力学数据,计算出MEDI-565的安全起始剂量,为后续的I期临床研究奠定了基础。
3.3 EpCAM靶向TCBs和CEA靶向TCBs
该研究讨论了T细胞接合双特异性抗体(TCBs)在固体瘤治疗中的应用,这些抗体可引起I期试验中的腹泻,表明存在非肿瘤肠道反应。
- 非临床安全性评估方法:在非临床开发中,动物模型未能预测EpCAM靶向或CEA靶向TCBs以及CEA靶向嵌合抗原受体(CAR)T细胞的肠道风险。这可能是由于不同物种间的免疫学差异和靶点组织相关表达的差异。研究人员探讨了患者源性肠类器官是否可以用于模拟EpCAM靶向TCBs的临床毒性,并对正在开发的CEA靶向TCBs的毒性进行建模。
- 研究结果与意义:该模型显示,所有EpCAM靶向分子触发了肠类器官中时间-和浓度-依赖性的诱导凋亡。这些结果与临床报告一致,表明EpCAM靶向双特异性抗体比结合CEA的抗体更频繁、更严重地引起肠道不良事件。这表明人类类器官可以提供一个稳健和敏感的系统来模拟健康器官中的TCB介导的毒性,优于未能预测这些肠道风险的动物模型。
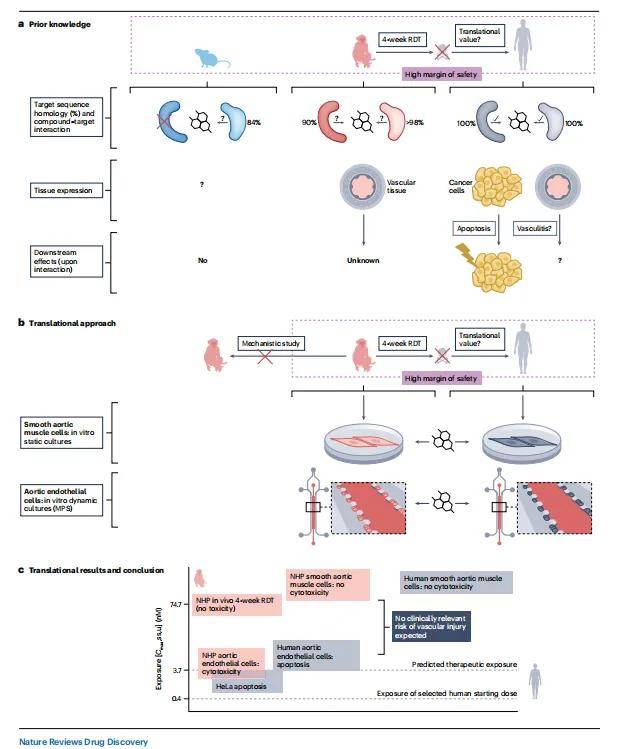
四、关键问题与答案
4.1. 为何新型疗法需要NAMs?
答案:新型疗法如双特异性抗体、基因治疗等,其靶点具有显著的人类特异性。以双特异性抗体为例,像MEDI-565这种CEA/CD3双抗,在猴体内由于不结合猴CD3,使得传统基于猴等动物模型的研究无法对其靶向毒性进行有效评估。从分子机制层面来看,动物(如啮齿类、非人类灵长类)的基因序列及蛋白表达与人类存在差异,导致许多人类特异性靶点在动物体内缺乏表达,或者即便有表达,其功能也与人类靶点大相径庭。这就使得传统动物模型难以模拟新型疗法在人体中的真实作用情况。
在实际研发过程中,以IMCgp100这种TCR融合蛋白药物开发为例,由于缺乏相关动物模型,研发团队采用了纯体外评估方案。通过将人肿瘤细胞与人免疫细胞进行共培养,模拟人体免疫系统对肿瘤细胞的攻击过程,同时开展细胞因子释放试验,监测免疫反应过程中细胞因子的释放情况。基于这些体外试验数据,成功确定了最小生物学效应剂量(MABEL),为后续的临床开发提供了关键的剂量参考依据,有力地推动了该药物从实验室走向临床应用的进程。
Kirkstall Quasi Vivo类器官串联器官芯片共培养系统,通过动态培养与多器官互作模拟,正在重塑疾病研究、药物开发与再生医学的范式。未来,随着技术迭代与跨领域合作深化,这一平台有望成为精准医学从实验室到临床的桥梁,推动人类健康事业的跨越式发展。
4.2. NAMs如何提升肝毒性预测?
答案:传统动物模型在预测人类肝毒性方面存在较大局限性。从胆汁酸代谢角度分析,人类胆汁酸组成以富含疏水胆汁酸为主,而大鼠、猴等动物的胆汁酸则以极性胆汁酸居多。这种胆汁酸组成的差异,使得动物模型难以准确模拟人类肝脏对药物的代谢及毒性反应过程。
NAMs中的3D肝类器官和肝芯片技术在提升肝毒性预测准确性方面具有显著优势。3D肝类器官通过模拟人体肝脏的三维组织结构,能够在体外重现肝脏细胞的功能及细胞间相互作用。在肝芯片中,构建了包含多种肝脏细胞类型(如肝细胞、肝星状细胞、内皮细胞等)的微流控芯片系统,模拟肝脏的微环境,包括血流、营养物质运输及代谢产物排泄等过程。通过这些技术,可以实时监测药物在类器官或芯片中的代谢过程及对肝脏细胞的毒性影响。
结合DILIsym计算模型,能够进一步深入解析药物导致肝毒性的机制。例如在BAY 1128688的研究中,动物试验未显示出肝毒性,但利用肝芯片进行研究时,发现该药物能够抑制BSEP(一种胆汁酸盐输出泵)的功能。BSEP功能受抑制后,胆汁酸无法正常排出肝细胞,导致胆汁酸在肝细胞内淤积,进而引发肝细胞损伤,这与临床中观察到的肝损伤症状高度一致。这种多技术联合的方式,从细胞、组织及系统层面全面解析药物肝毒性机制,大大提升了肝毒性预测的准确性和可靠性。
4.3. 监管层面如何推动NAMs应用?
答案:监管机构通过一系列立法与指南大力支持NAMs的应用与发展。
- FDA现代化法案2.0/3.0:该法案具有重要的里程碑意义,明确允许在药物研发过程中使用细胞类器官模型、计算工具等替代动物试验。以IMCgp100的研发为例,FDA批准了其采用纯体外安全评估方案,这一举措为新型疗法的研发开辟了新路径。法案的实施激励了药企积极探索和应用NAMs技术,因为使用这些新技术不仅能够满足监管要求,还能在一定程度上缩短药物研发周期、降低研发成本,同时提升药物安全性评估的准确性。
- EMA指南:EMA指南明确规定,当动物模型无法有效评估药物安全性时,必须采用人体体外模型进行补充评估。例如在转基因动物替代试验中,要求企业充分考虑使用人类细胞模型、类器官等NAMs技术。此外,EMA还鼓励药企在药物研发早期阶段就与监管机构进行沟通,详细阐述所采用的NAMs方案,以便监管机构能够及时给予指导和建议,确保研发过程符合法规要求,促进NAMs技术在药物研发中的合理应用。
- OECD标准:OECD发布的皮肤致敏性评估的“定义方法”(DA),为药物领域中NAMs的标准化应用提供了重要参考。该标准详细规定了相关测试的流程、方法及评估指标,使得不同实验室、不同研究团队在开展相关研究时能够遵循统一的规范,提高了研究结果的可比性和可靠性。这对于推动NAMs在药物安全性评估等领域的广泛应用具有积极的促进作用,有助于建立全球范围内对NAMs技术的信任和认可。
北京基尔比生物科技公司主营产品:
Kilby 3D-clinostat 旋转细胞培养仪,Kilby Gravite微/超重力三维细胞培养系统,
3D回转重力环境模拟系统,随机定位仪,
类器官芯片摇摆灌注仪,
Kirkstall 类器官串联芯片灌流仿生构建系统