

CD4+T经1α介导的内皮氧化酶1增加诱导HIV相关的内皮功能障碍和高血压
2025-12-10 来源:本站 点击次数:754CD4+ T Cells Expressing Viral Proteins Induce HIV-Associated Endothelial Dysfunction and Hypertension Through Interleukin 1α-Mediated Increases in Endothelial NADPH Oxidase 1
Keywords: CD4-positive T-lymphocytes; HIV; endothelium; hypertension; interleukin-1alpha; oxidative stress; viral proteins.
心血管疾病(CVD)现已成为HIV感染者(PLWH)的主要死因,而高血压是CVD的主要风险因素,在联合抗逆转录病毒疗法(cART)治疗下的PLWH中非常普遍。尽管HIV相关高血压的流行病学问题已经明确,但其病因仍然大多未知,且缺乏关于其潜在机制的实验研究。
较高的非传染性合并症数量已被证明不足以解释PLWH中较高的高血压患病率,这支持病毒学或治疗相关因素对病理生理学的贡献。有趣的是,接受cART治疗的PLWH 患者的高血压患病率始终高于cART初治患者,而cART初治患者则低于未感染者。这些观察表明,CD4+ T淋巴细胞(T细胞),在接受cART治疗的PLWH中含量高,在cART初治患者中含量低,可能与高血压有关。
点击了解:仿生多细胞动态共培养系统
CD4+ T细胞是cART治疗的PLWH中最大且最为明确的HIV储存库。HIV仍保持转录活性在CD4+ T细胞中,病毒蛋白表达依然存在。HIV衍生蛋白在cART治疗的PLWH体内循环并创造促进CVD的炎症环境。因此,可假设雄性和雌性小鼠中表达HIV衍生蛋白的CD4+ T细胞通过释放促炎细胞因子诱导高血压。为验证这一假设,美国南阿拉巴马大学莫比尔分校血管生物学中心及奥古斯塔大学乔治亚医学院生物化学和分子生物学系团队在一项研究中结合了骨髓移植(BMT)的Tg26 HIV小鼠模型与及T细胞过继性转移,并分析了PLWH中主动脉标本。研究成果发表于Circulation 期刊题为“CD4+ T Cells Expressing Viral Proteins Induce HIV-Associated Endothelial Dysfunction and Hypertension Through Interleukin 1α-Mediated Increases in Endothelial NADPH Oxidase 1 ”。

首先,为研究病毒蛋白在HIV相关高血压发病机制中的作用,使用Tg26转基因小鼠,提供了一种非传染性的HIV模型,类似于接受抗逆转录病毒治疗的PLWH 。实验报告显示,在正常雄性和雌性Tg26小鼠中病毒蛋白的表达在保持昼夜节律的同时,血压(BP)有所增加(图1 A、B、D、E),但不影响心率。基于血管系统在控制血压中的关键作用以及HIV对血管功能的已知有害影响,评估了二级肠系膜动脉的血管反应性。Tg26小鼠中病毒蛋白的表达导致雄性和雌性(图1 C、F)小鼠对乙酰胆碱(ACh)的血管舒张反应明显受损,而对硝普钠(SNP)的血管舒张反应则未改变,这支持内皮细胞水平的功能障碍。这些数据表明。Tg26小鼠中病毒蛋白的表达会提高血压并损害内皮依赖性血管舒张反应。
为了检验造血细胞衍生的病毒蛋白是否可能导致内皮功能障碍和高血压,采集并移植了WT小鼠和Tg26小鼠的骨髓(图1 G)。通过尾袖测量血压显示,相较于WT-WT,Tg26-WT小鼠的BMT显著提高了雄性和雌性小鼠的收缩压(SBP)(图1 H、I)。从WT到Tg26小鼠的BMT消除了上述雄性和雌性Tg26小鼠报告的高血压表型,并将血压水平降低至WT-WT组(图1 H、I)。值得注意的是,血管反应性研究的结果与BP数据相符。事实上,从Tg26到WT小鼠的BMT重现了正常Tg26小鼠中的肠系膜动脉内皮功能障碍,而从WT小鼠到Tg26小鼠的BMT则恢复了肠系膜动脉对ACh的舒张反应(图1 J、K)。在肠系膜动脉中观察到,从Tg26到WT小鼠的BMT损伤了主动脉内皮功能,而从WT到Tg26小鼠的BMT则恢复了导管动脉对ACh的反应而不影响SNP介导的舒张反应。这些结果表明,源自造血细胞的病毒蛋白是雄性和雌性Tg26小鼠内皮功能障碍和高血压的根源。

图1 雄性和雌性小鼠的造血细胞中HIV蛋白的表达会增加血压,并损害阻力血管的内皮功能。
研究人员试图确定导致内皮功能障碍和高血压的免疫细胞亚型,因此开发了一种新系统(图2 A),将WT胸主动脉与脾脏来源的CD3+ T细胞或BM来源巨噬细胞共培养。与暴露于WT CD3+ T细胞相比,WT主动脉暴露于Tg26 CD3+ T细胞会损害血管舒张反应(图2 B),但暴露于来自WT或Tg26小鼠的BM来源巨噬细胞并未改变血管舒张反应(图2 C)。
基于T细胞参与高血压需要共刺激的证据,通过用CTLA4-Ig(如abatacept)阻断共刺激信号,进一步研究了T细胞对HIV相关高血压发病机制的贡献。阿巴西普(Abatacept )降低了平均动脉压(图2 D、E)和舒张压,同时恢复了阻力性(图2 F)和传导性血管的内皮功能,但未影响平滑肌细胞依赖性血管舒张(图2 G)。这些数据综合提供了体外和体内证据,表明源自T细胞的病毒蛋白对观察到的BP升高和内皮功能障碍有影响。
使用CD4+ 辅助T细胞和CD8+ 细胞毒性T细胞重复共培养实验(图2 A)以缩小参与的T细胞亚型,发现暴露于CD4+,而非CD8+,会损害内皮依赖性舒张反应。进一步测试CD4+ T细胞的作用,发现CD4+ 耗竭使Tg26小鼠的SBP水平降至WT小鼠水平,且未影响WT小鼠的血压,同时也恢复了对ACh的血管舒张反应,但未改变血管平滑肌细胞依赖性血管舒张。在缺失CD4+ 和 CD8+ T细胞的TCRα敲除小鼠中进行CD4+ T细胞的过继性转移,发现相较于从WT小鼠分离的CD4+ T细胞,Tg26 CD4+ T细胞的过继性转移显著升高了SBP。这些结果共同支持,CD4+ T细胞表达的病毒蛋白在高血压和内皮功能障碍中的作用。
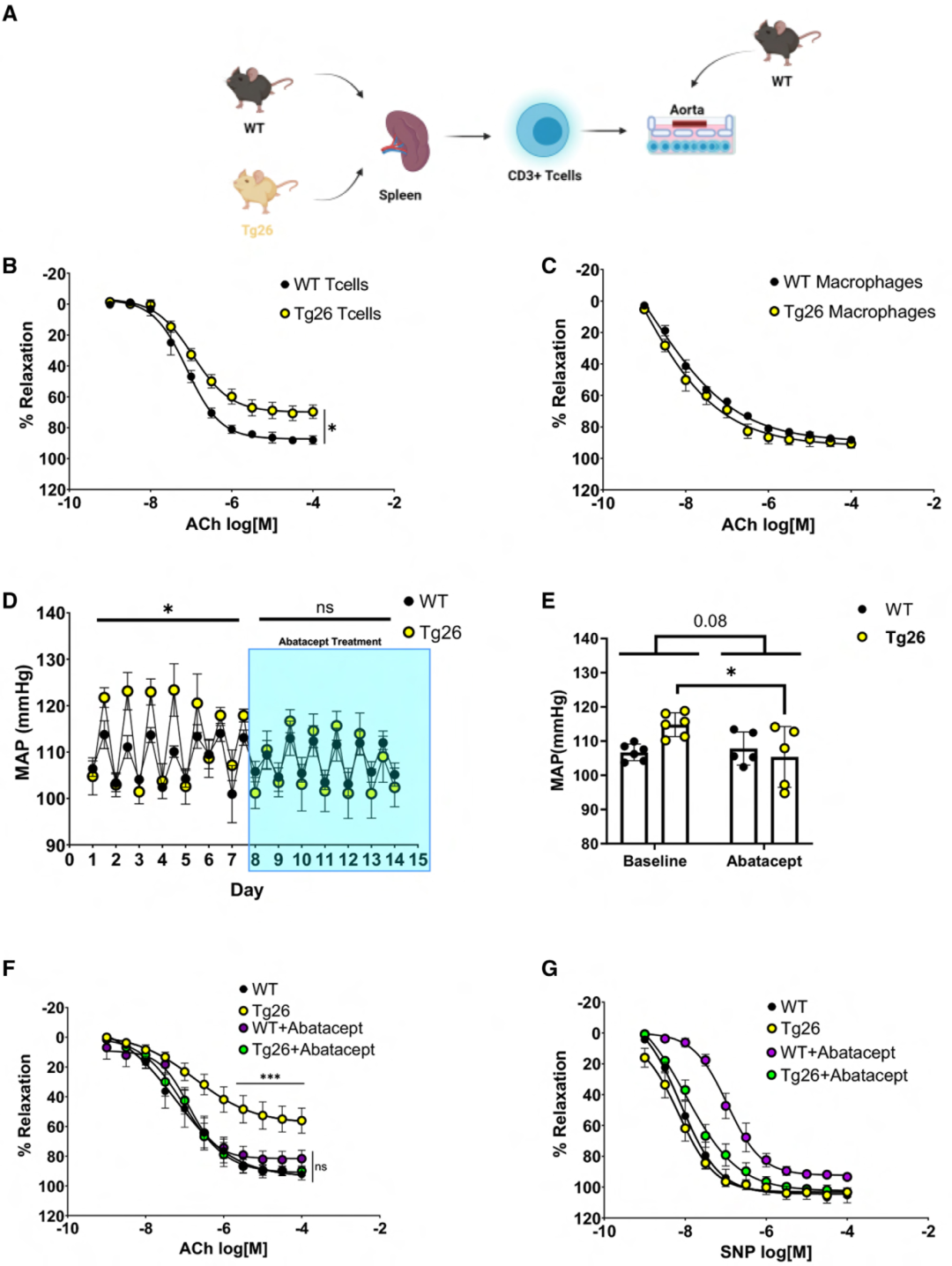
图2 CD3+ T细胞中HIV蛋白的表达可诱发内皮功能障碍和高血压。
基于促炎细胞因子在高血压发展中的作用,接下来对完整和BMT小鼠的血浆进行了Cytokine Panel分析,测量显示,Tg26小鼠的病毒蛋白表达显著提高了循环中的IL-1α水平(图3 A、B),而其他促炎细胞因子则无显著变化。从Tg26到WT小鼠的BMT增加,用WTs替代Tg26 BM降低IL-1α水平(图3 C、D)。此外,较CD8+ T细胞,病毒蛋白表达在CD4+ T细胞中诱导IL-1α更显著地增加(图3 E、F)。阿巴西普抑制T细胞共刺激(图3 G)和CD4+ T细胞耗竭(图3 G)降低了Tg26小鼠血浆IL-1α水平,表明CD4+ T细胞是循环IL-1α升高的来源。
正常WT和Tg26小鼠接受IL-1α中和抗体处理,发现IL-1α中和使Tg26 小鼠BP水平降低(图3 H、I),心率无变化,且恢复阻力动脉内皮依赖性舒张(图3 J),但未影响平滑肌细胞依赖性血管舒张。同时,在IL-1α中和抗体存在下重复了T细胞-主动脉共培养实验,发现 IL-1α中和保护了主动脉环免受Tg26 CD4+ T细胞诱导的内皮功能障碍(图3 K),且未影响SNP介导的舒张。将WT小鼠接受每日20 mg/kg的外源性IL-1α治疗,发现IL-1α注射升高了SBP,并损害了内皮依赖性血管舒张(图3 L、M),但平滑肌细胞依赖性舒张无异。这些数据支持,CD4+ T细胞来源的IL-1α在HIV病毒蛋白相关的内皮功能障碍和高血压中的作用。
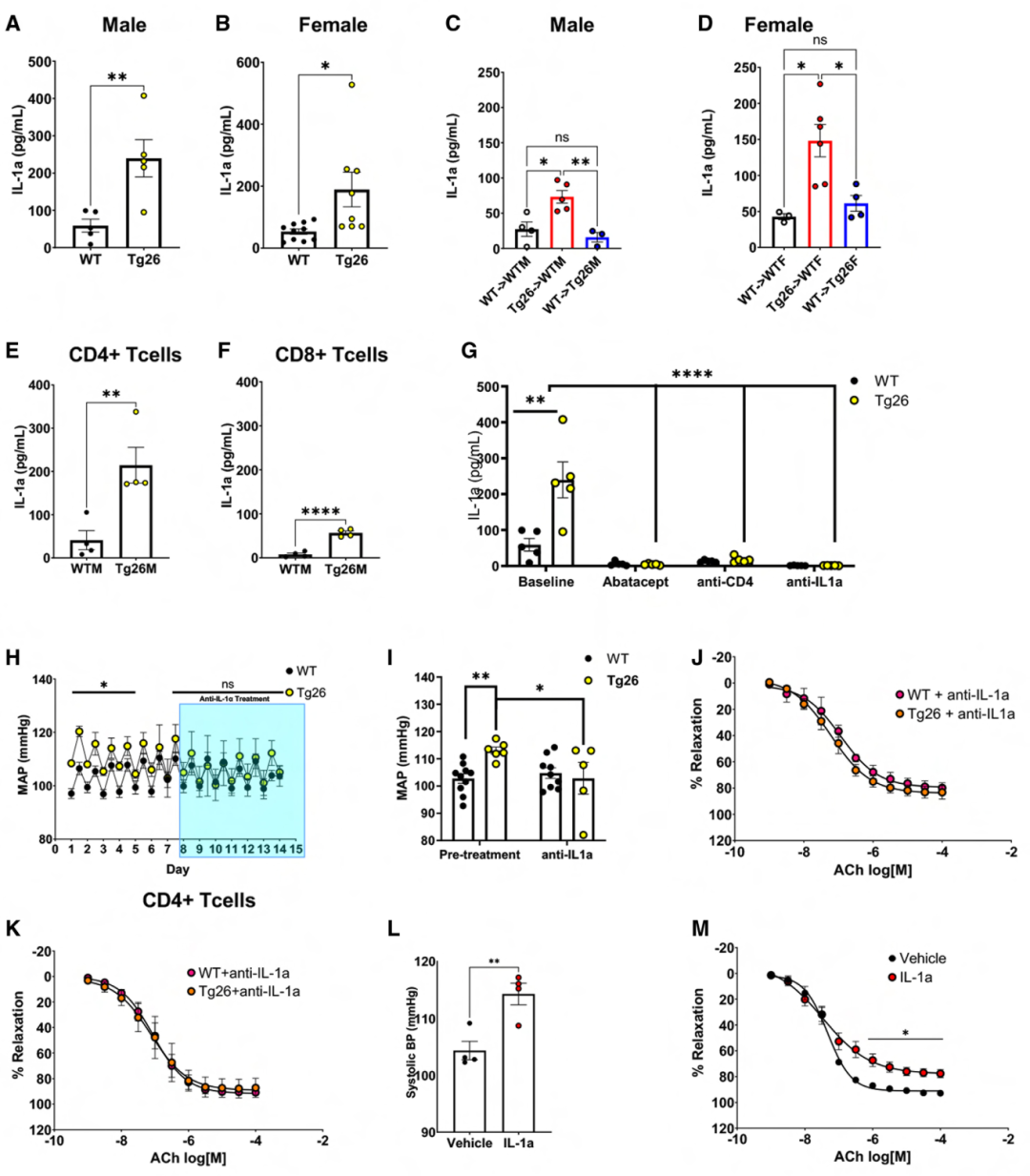
图3 CD4+ T细胞中病毒蛋白的表达会诱导IL-1α的表达,从而损害内皮功能并诱发高血压。
过量的活性氧,尤其是高NAD(P)H氧化酶衍生的活性氧,与HIV和T细胞介导的高血压和血管功能障碍有关。因此,实验定量了主动脉中NOX亚型转录物表达,发现 Tg26小鼠的病毒蛋白表达增加了NOX1,但非NOX2和NOX4的显著增加,CD4+ 耗竭降低了完整小鼠主动脉中的NOX1水平,NOX2和NOX4水平未显著变化。与上述发现一致,从Tg26到WT小鼠的BMT仅增加了主动脉中的NOX1,且WT大主动脉暴露于Tg26 CD3+ T细胞仅增加NOX1。为验证这些发现的可转化性,测量了接受心脏搭桥手术的PLWH丢弃的主动脉活检标本中NOX的表达,发现PLWH患者的NOX1水平高于血清阴性患者。使用选择性NOX1抑制剂GKT771,发现其消除了WT与Tg26雄性小鼠之间的血管舒张差异,也消除了WT-WT及Tg26-WT BMT小鼠之间的差异。NOX1的缺失可防止病毒蛋白诱导的高血压和内皮功能障碍,同时未改变血管平滑肌细胞依赖性舒张。这些数据表明,CD4+ 来源的病毒蛋白通过NOX1依赖机制损害内皮功能并升高BP。
研究人员假设IL-1α会增加内皮NOX1表达。为验证这一假设,将hMVECs暴露于IL-1α,发现 IL-1α增加了内皮NOX1表达(图4 A),但NOX2、NOX4或NOX5(图4 B-D)表达无显著变化。此外,测量了8-羟基-2'-脱氧鸟苷水平(8-OHdG,活性氧诱导氧化损伤的指标),发现IL-1α显著提高了hMVECs中的 8-OHdG水平(图4 E),表明 IL-1α是NOX1表达和活性氧产生增加的来源。使用NOX1抑制剂GKT771,发现抑制 NOX1消除了IL-1α处理小鼠与载体处理小鼠肠系膜血管舒张差异(图4 F)。为探究IL-1α促进内皮NOX1表达的潜在机制,测试了NF-κB和caspase 1的作用。发现抑制 NF-κB和caspase 1均能防止IL-1α诱导的内皮依赖性舒张受损(图4 G)以及IL-1α介导的内皮NOX1增加(图4 H、I)。这表明,IL-1α会增加hMVECS中的NOX1和氧化应激。
最后,为确定CD4+ T细胞是否能介导NOX1增加,将接受心脏搭桥手术的血清阴性患者丢弃的人类主动脉标本暴露于WT或Tg26 CD4+ T细胞条件培养基(图4 J),发现暴露于CD4+ T细胞培养基会增加NOX1表达,并诱导NOX2上升的趋势,但未达到统计显著性(图4 K-N)。这些结果表明,源自CD4+ T细胞的病毒蛋白会增加人体血管NOX1表达。
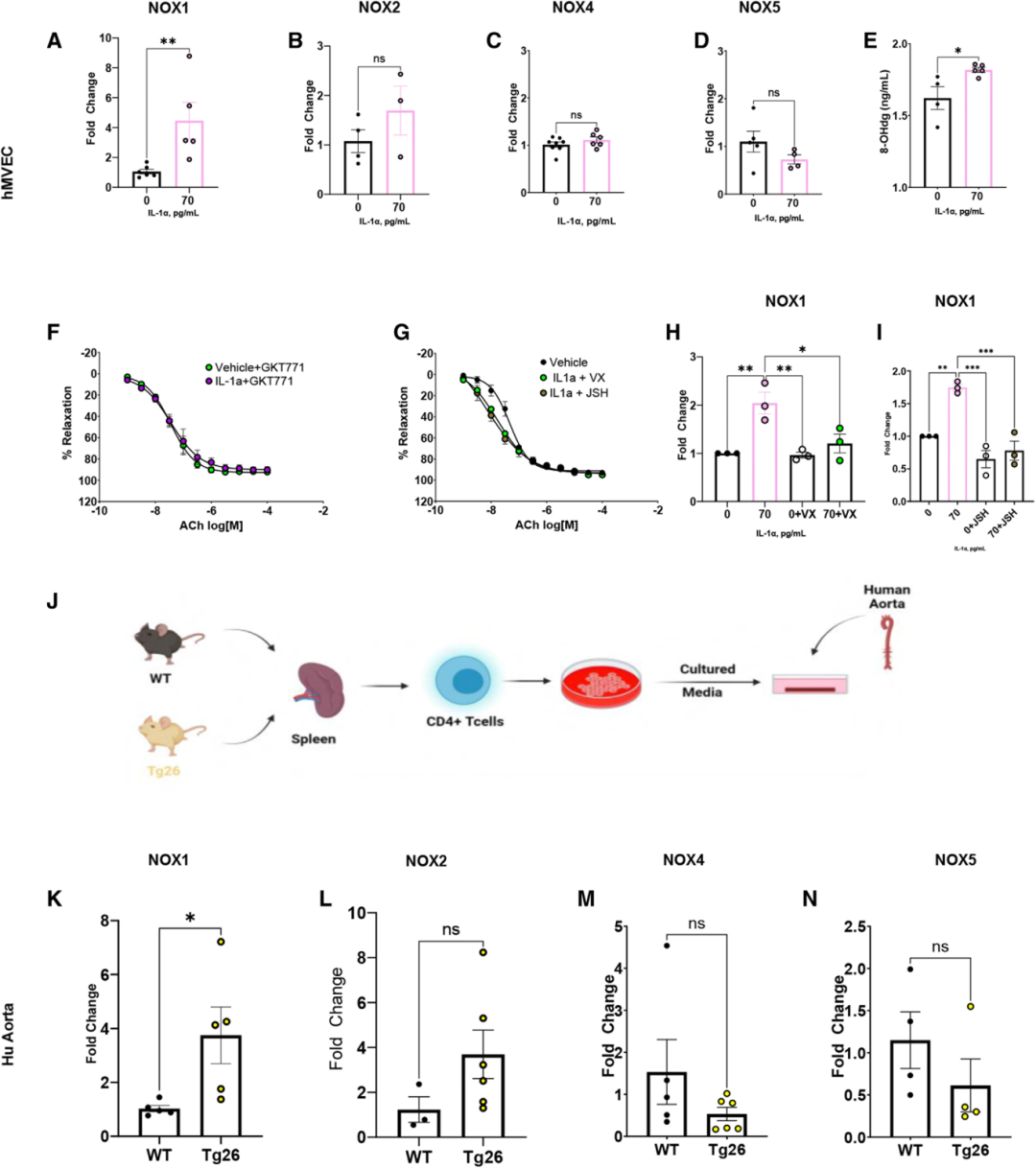
图4 IL-1α和CD4+ T细胞增加内皮NOX1和活性氧的水平。
总之,该研究shouci提供了实验证据,表明CD4+ T细胞中病毒蛋白的表达会导致促炎细胞因子IL-1α的分泌,从而提高内皮细胞的NOX1水平,进而损害内皮舒张功能并提高血压。这是shouge机制性研究介绍了NOX1抑制剂、IL-1α抗体和IL-1受体阻断,作为缓解PLWH 患者高血压患病率上升的潜在治疗途径。
参考文献:Kress TC, Barris CT, Kovacs L, Khakina BN, Jordan CR, Bruder-Nascimento T, Stepp DW, MacArthur R, Patel VS, Chen J, Pacholczyk R, Kennard S, Belin de Chantemèle EJ. CD4+ T Cells Expressing Viral Proteins Induce HIV-Associated Endothelial Dysfunction and Hypertension Through Interleukin 1α-Mediated Increases in Endothelial NADPH Oxidase 1. Circulation. 2025 Apr 22;151(16):1187-1203. doi: 10.1161/CIRCULATIONAHA.124.070538. Epub 2025 Feb 5. PMID: 39907014; PMCID: PMC12011537.
原文链接:https://pubmed.ncbi.nlm.nih.gov/39907014/
图片来源:所有图片均来源于参考文献
小编旨在分享、学习、交流生物科学等领域的研究进展。如有侵权或引文不当请联系小编修正。如有任何的想法以及建议,欢迎联系小编。感谢各位的浏览以及关注!进入官网www.naturethink.com或关注“Naturethink”公众号,了解更多相关内容。
点击了解:仿生多细胞动态共培养系统