

如何从数据库挖掘基因并筛选 TagSNP(医学篇)
2024-08-04 来源:公众号 点击次数:2879在人医学遗传学研究中,SNP 与疾病相关性一直是广泛的研究课题。很多人在刚开始接触课题,在没有前期研究基础指示的目的基因时,都会选择从公共数据库中寻找与疾病相关的基因或者SNP进行研究。本文小编带你学习如何从数据库中挖掘基因,并聚焦疾病相关的重要通路。
技术路线
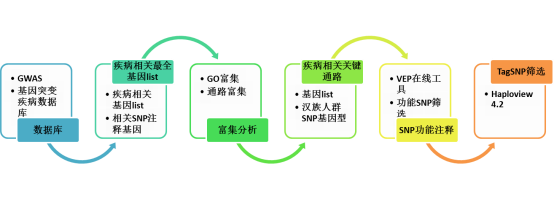
1.疾病/复杂性状相关数据库
百度搜索可以获得很多人疾病相关数据库的使用说明,在此不再赘述。我们常用的人类疾病数据库是 DisGeNET(https://www.disgenet.org/search ),常用 GWAS 数据库是 EMBL-EBI 的 GWAS catalog(https://www.ebi.ac.uk/gwas/ )。
2.疾病相关最全基因 list
通过数据库下载疾病相关基因列表;相关 SNP 也可以在 VEP 在线注释工具(http://asia.ensembl.org/Multi/Tools/VEP )注释其所在的基因。将两部分基因合并,获得基本相关较为全面的基因列表。
3.富集分析----聚焦疾病/复杂性状相关通路
富集分析使用 R 语言的 clusterProfiler 程序包。即使不会 R 语言,不懂编程,一样可以完成分析。
安装 R
百度搜索 R,找到合适的下载源;也可直接点击链接https://cran.dcc.uchile.cl/,选择合适的版本下载。
#设置工作目录
运行 R 后,在《文件》菜单下选择《改变工作目录》;
将基因名替换好以后,直接复制下列代码到 R,回车运行即可,除此之外可以不做任何改动。如果已经安装 clusterProfiler 程序包,请从基因编号转换开始。
#安装 clusterProfiler 程序包,此种安装方法适合 R3.5.2 及以下版本,R3.6.0 以上版本请参考文后补充说明。
source("https://bioconductor.org/biocLite.R")
biocLite("clusterProfiler")
#安装 KEGG.db
biocLite("kegg.db")
#安装人 org.db 数据库
biocLite(“org.Hs.eg.db”)
#基因编号转换
#将基因名称列表复制给(或者任何你喜欢的文件名),如需分析自己特定的基因集,可替换括号内容,每个基因名称,用””,隔开。
library(clusterProfiler)
yh <- c("GPX3", "GLRX", "LBP", "CRYAB", "DEFB1", "HCLS1", "SOD2", "HSPA2",
"ORM1", "IGFBP1", "PTHLH", "GPC3", "IGFBP3","TOB1", "MITF", "NDRG1", "NR1H4", "FGFR3", "PVR", "IL6", "PTPRM", "ERBB2", "NID2", "LAMB1", "COMP", "PLS3", "MCAM", "SPP1", "LAMC1", "COL4A2", "COL4A1", "MYOC", "ANXA4", "TFPI2", "CST6", "SLPI", "TIMP2", "CPM", "GGT1", "NNMT", "MAL", "EEF1A2", "HGD", "TCN2", "CDA", "PCCA", "CRYM", "PDXK", "STC1", "WARS", "HMOX1", "FXYD2", "RBP4", "SLC6A12", "KDELR3", "ITM2B")
#利用 cluterProfiler 内置的 bitr 函数进行基因编号转换,并将转换后的信息存储在 gene 文件中
gene <- bitr(yh, fromType="SYMBOL", toType=c("ENTREZID"), OrgDb="org.Hs.eg.db")
head(gene)
##提取 gene 数据中的 ENTREZID 列,并赋值给 DE_list
DE_list <- gene$ENTREZID
#去除重复值
DE_list[duplicated(DE_list)]
integer(0)
#调用 org.Hs.eg.db,并查看文件的各列名称信息
library(org.Hs.eg.db)
columns(org.Hs.eg.db)
#GO_MF 富集,基于基因数目,如果使用的是个人电脑,配置不高,为防止程序卡死,建议 MF\CC\BP 单个来运行,生成的图片也逐个生成保存后再运行下一个。
MF <- enrichGO(gene = DE_list, #差异基因 vector
keyType ="ENTREZID",
OrgDb = org.Hs.eg.db, #对应的OrgDb
ont = "MF", #GO 分类名称,CC BP MF
pAdjustMethod = "BH", #Pvalue 矫正方法
pvalueCutoff = 0.05, #Pvalue 阈值
qvalueCutoff = 0.05, #qvalue 阈值
readable = TRUE) #TRUE 则展示SYMBOL,FALSE 则展示原来的ID
#将 MF 对象转换为 dataframe,新版本可以用 as.data.frame(MF)
MF_results<-summary(MF)
#生成 barplot PDF 格式,x 轴为 GeneRatio,展示前 20 富集的 GO,数字可以调整
pdf(file = "MF_barplot.pdf")
barplot(MF, showCategory=20, x = "GeneRatio")
dev.off()
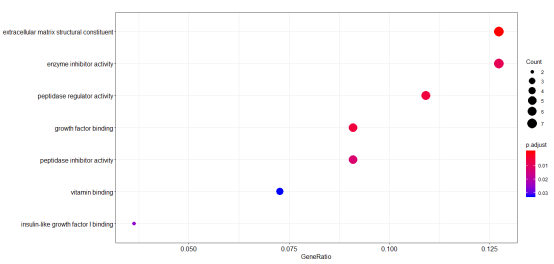
#生成 MF 气泡图
dotplot(MF)
#GO_CC 富集,基于基因数目
CC <- enrichGO(gene = DE_list, #差异基因 vector
keyType ="ENTREZID",
OrgDb = org.Hs.eg.db, #对应的OrgDb
ont = "CC", #GO 分类名称,CC BP MF
pAdjustMethod = "BH", #Pvalue 矫正方法
pvalueCutoff = 0.05, #Pvalue 阈值
qvalueCutoff = 0.05, #qvalue 阈值
readable = TRUE) #TRUE 则展示SYMBOL,FALSE 则展示原来的ID
#将 CC 对象转换为 dataframe,新版本可以用 as.data.frame(CC)
CC_results<-summary(CC)
#生成 barplot PDF 格式,x 轴为 GeneRatio,展示前 20 富集的 GO,数字可调整
pdf(file = "CC_barplot.pdf")
barplot(CC, showCategory=20, x = "GeneRatio")
dev.off()
#生成 CC 气泡图
dotplot(CC)
#GO_BP 富集,基于基因数目
BP <- enrichGO(gene = DE_list, #差异基因 vector
keyType ="ENTREZID",
OrgDb = org.Hs.eg.db, #对应的OrgDb
ont = "BP", #GO 分类名称,CC BP MF
pAdjustMethod = "BH", #Pvalue 矫正方法
pvalueCutoff = 0.05, #Pvalue 阈值
qvalueCutoff = 0.05, #qvalue 阈值
readable = TRUE) #TRUE 则展示SYMBOL,FALSE 则展示原来的ID
#将 BP 对象转换为 dataframe,新版本可以用 as.data.frame(BP)
BP_results<-summary(BP)
#生成 barplot PDF 格式,x 轴为 GeneRatio,展示前 20 富集的 GO,数字可调整
pdf(file = "BP_barplot.pdf")
barplot(BP, showCategory=20, x = "GeneRatio")
dev.off()
#生成 BP 气泡图
dotplot(BP)
#KEGG pathway 富集
ekp <- enrichKEGG(gene = DE_list,
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 0.05)
ekp_results <- summary(ekp)
#生成 KEGG 富集分析的 barplot 图,数字可调整
barplot(ekp, showCategory=20, x = "GeneRatio")
#生成气泡图
dotplot(ekp)
#基因和富集排名第 1 的pathway对应关系
cnetplot(ekp, showCategory = 1)
#输出 pathway 富集结果,可以用 excel 打开查看
write.table(ekp, file = "ekp.txt",
sep = "\t", quote = F, row.names = T)
#查看通路
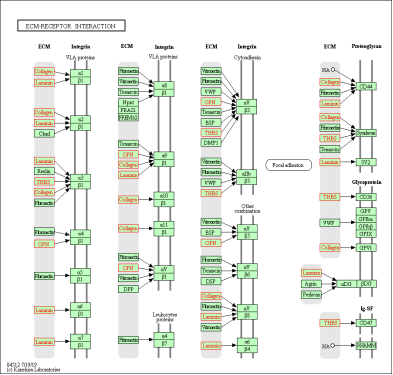
browseKEGG(ekp,'hsa04512')
过程文件展示
MF_barplot
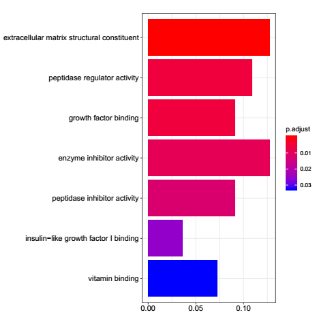
MF_dotplot
KEGG_barplot
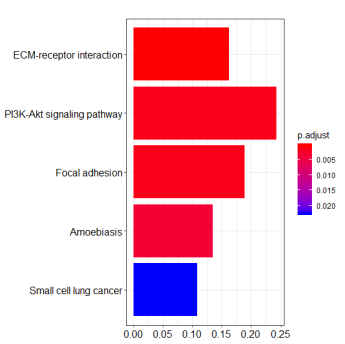
KEGG_dotplot
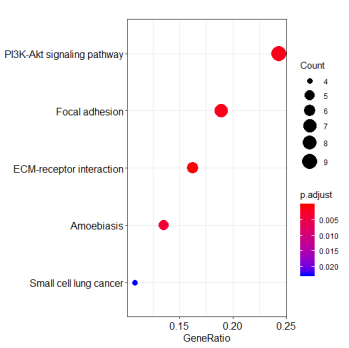
KEGG enrichment pathway browse
补充说明
#R3.6.0 以上版本安装方法不同于 R3.5.2 及以下版本,biocManager 安装方法如下:
If(!requireNamespace(「BiocManager」,quietly=TRUE))
Install.packages(「BiocManager」)
BiocManager::install(「clusterProfiler」,version = 「3.8」)
相关文章
更多 >