

低多样性文库在NextSeq500/550和MiniSeq上测序及Index混合指导方针
2022-04-13 来源:Illumina因美纳公众号 点击次数:2420
如何在NextSeq™ 500/550和MiniSeq™平台进行低多样性文库测序
若希望低多样性文库在NextSeq™ 500/550和MiniSeq™平台上获得精确和稳定的测序结果,需要精心设计的实验以及良好的生信分析。低多样性文库最常见的例子是基于扩增方法制备的文库,例如16S宏基因组文库。这些文库扩增引物结合的位置,即起始位点的DNA序列基本是相同的。单一的位点会导致碱基组成不平衡,以至于碱基组成从一个循环到下一个循环可能会发生剧烈变化。
NextSeq™ 500/550和MiniSeq™系统使用的是双通道测序技术。因此,确保每个循环中均存在4种DNA碱基是非常重要的,这样分析软件才能正确鉴定DNA簇并精确读取碱基序列。低多样性文库测序时,为了满足这种要求,我们建议在设计实验时,采用以下几种方法保证每个循环的多样性。
利用标签区分技术,在测序中加入多个带有标签的来自多种应用的样品, 关于NextSeq™ 500/550和MiniSeq™测序系统上Index混合的指导方针我们会在下面详细介绍。
· 用多样性较高应用的带有标签的样品,例如全基因组测序文库,来增加文库碱基多样性
· 为了获得最佳测序结果,可以利用单标签或者双标签技术,将多个样品在同一张Flow Cell上进行测序
在测序时掺入全基因组样品(例如PhiX)
· 可以加入50% PhiX为初始掺入比例,然后根据一级分析和二级分析的结果,逐步降低PhiX掺入比例。掺入这样的样品能提供每个循环必需的碱基多样性。
在NextSeq™ 500/550和MiniSeq™平台原始簇密度最佳范围如下所示, 而低多样性文库在此基础上须视情况降低上样量以提升测序质量:
· MiniSeq™试剂原始簇密度最佳范围为170-220 K/mm2
· NextSeq™ 500/550试剂原始簇密度最佳范围为170-220 K/mm2
NextSeq™ 500/550和MiniSeq™测序系统Index混合的指导方针
在NextSeq™ 500/550 和MiniSeq™测序系统上对混合文库进行Index测序时,非常重要的一点是要选择合适的Index组合,否则可能会因为读Index时簇定位失败而导致Index测序失败。
NextSeq™ 500/550和MiniSeq™为双通道测序
NextSeq™ 500/550和MiniSeq™测序系统只需要2张图片就可以区分4种碱基:一张图片来自红光通道,另一张图片来自绿光通道。不同于每种碱基各带一种单独的荧光标记的四通道测序,双通道测序使用两种荧光染料,C碱基标记红色,T碱基标记绿色,A碱基同时标记红色和绿色,G碱基没有荧光标记。
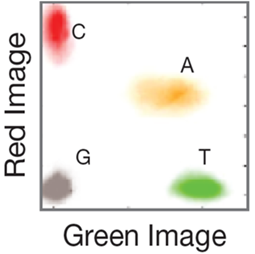
图1:双通道SBS荧光成像(假彩色图片仅是效果图)为了提高4种DNA碱基的检测速度,MiniSeq™和NextSeq™ 500/550只用了2个图像分别捕捉红色和绿色波段的信号。仅在红色或绿色图像检测到的cluster分别被转译为C和T碱基。同时在红色图像和绿色图像被检测到的cluster被转译为A碱基,在两张图片中都没有检测到信号的cluster将被认为是G碱基。
NextSeq™500/550和MiniSeq™相关Index的考量
Index reads中前两个循环必须至少有一个碱基不是G。如果Index read前面两个碱基都是G,就会由于没有荧光信号导致cluster定位失败,所以Index的前两个循环中必须有一个碱基有信号才能确保Index的正常拆分。
选择Index进行组合时,每个循环都至少有一个通道有信号,最好是两个通道都有信号。这样碱基读取的过程才能确保数据分析的精确。
· 红色通道对应A或者C碱基
· 绿色通道对应A或者T碱基
Index组合的示例
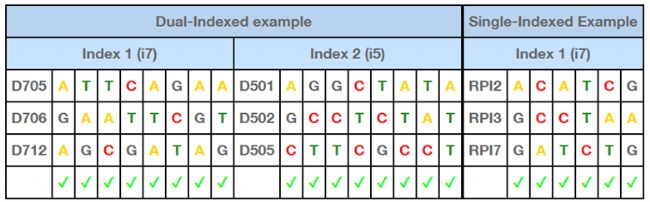
理想的Index组合:每个循环需要两个通道都有信号
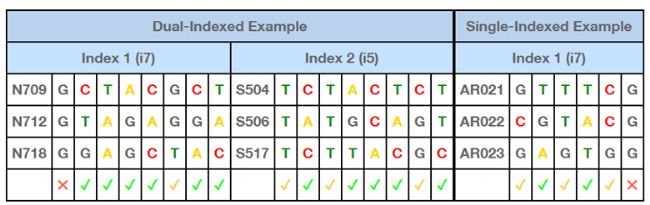
可接受的Index组合:只有一个通道有信号,但是仍然有足够的信号进行测序
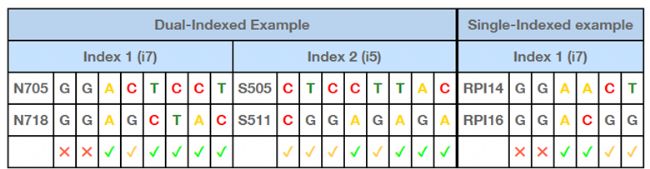
不好的Index组合:Index read前两个碱基都是G,没有信号产生,导致cluster定位失败。

上述中的这些建议能够使得NextSeq™ 500/550和MiniSeq™平台进行低多样性文库测序。