

反向找靶案例之中药单体和靶点的结合模式预测及验证
2024-09-19 来源:本站 点击次数:1970
之前,小陶介绍了一种反向靶点发现算法——COMET,并使用了 Apiopaeonoside 和 Sciadopitysin 两种中药单体进行了操作演示。

接下来,让我们一起回顾一下之前的实验结果,并探讨后续推荐的实验思路。
结果分析思路
Apiopaeonoside 反向找靶的结果如下图所示:
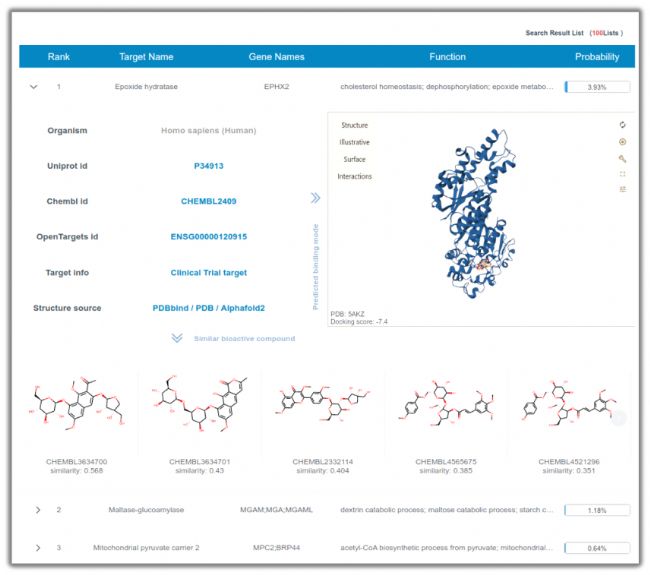
假设本次实验发现 Apiopaeonoside 能治疗慢性阻塞性肺疾病、高血压,而预测的第一个靶点 Epoxide hydrolase 刚好和这两种疾病相关,所以,第一个靶点虽然打分(Probability)较低,但是,其仍然值得我们尝试后续的实验。
Sciadopitysin反向找靶结果如下图所示:
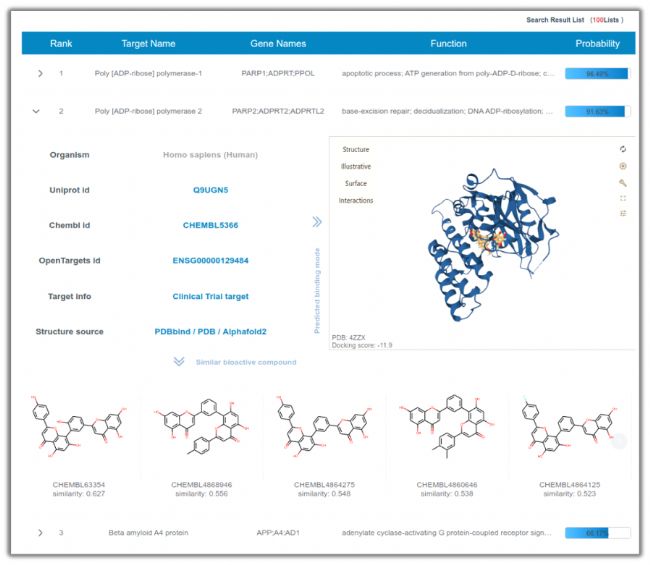
假设 Sciadopitysin 有治疗卵巢癌的效果,那么 Poly [ADP-ribose] polymerase 2 打分虽然不如 Poly [ADP-ribose] polymerase-1 好,但是,Poly [ADP-ribose] polymerase 2 和卵巢癌息息相关,所以,Poly [ADP-ribose] polymerase 2 仍然是我们首先考虑的靶点。
后续实验验证
根据假设,目前已经得到了两个分子可能的靶点。后续一般可以用 分子亲合力实验 来证明小分子和蛋白确实是有结合的,比如SPR(表面等离子体共振技术)、MST(微量热泳动技术)。实验需要用到纯化蛋白和小分子单体及对应的仪器,蛋白和小分子陶术生物都可以提供,如果没有实验仪器,也可以找陶术生物来做验证实验!
除上述说的湿实验验证方法外,也可以尝试采用动力学作为补充实验。如黄以超团队在“Environmentally realistic dose of tire-derived metabolite 6PPD-Q exposure causes intestinal jejunum and ileum damage in mice via cannabinoid receptor-activated inflammation ”中所做实验一样。
分子动力学模拟
本次实验将以 Sciadopitysin 和 Poly [ADP-ribose] polymerase 2 复合物结构为演示,采用 Amber24/Ambertool24 进行分子动力学模拟实验。本次实验细节内容将在陶术小课堂中展示。内容较多,会分步介绍,欢迎扫码关注~
 蛋白体系的准备
蛋白体系的准备
首先,我们对得到的结构进行调整,我们需要注意以下7点:
A Non-Standard Residues
B Metals
C Experimental methods noted in the paper associated with the PDB
D Solvent molecules or crystallization buffer
E Missing electron density (amino acids)
F Disulfide Bonds
G Protonation States
A Non-Standard Residues
Non-Standard Residues(非标准残基)是指PDB中20种天然氨基酸以外的其它氨基酸或者分子,包括辅因子(NADH、血红素等)、非标准氨基酸(羟脯氨酸等)、抑制剂/激动剂或底物。如果有任何非标准氨基酸,我们应该考虑如何对这些残基进行建模。对于不含金属或类金属的小有机分子,构建其结构和使用antechamber一样简单。对于更复杂的非标准残基,需要使用别的方法。
B Metals
金属的处理比较复杂,如果复合物结构中包含金属,应该仔细考虑如何对其建模。可以参考amber软件手册。或者在以下两个位置查看软件目前已有的金属力场$AMBERHOME/dat/leap/lib 和 $AMBERHOME/dat/leap/parm。
C Experimental methods noted in the paper associated with the PDB
使用PDB晶体结构的时候,一定要好好阅读一下对应的文献,并注意任何重要的结构特征(比如二硫键)。还要注意用于解析结构的实验程序。有任何问题,都可以直接编辑PDB文件来修复。注意以下几点:
在某些情况下,溶剂或结晶缓冲液可能与蛋白质一起结晶,并包含在PDB文件中。因为我们后续采用显式溶剂体系,所以我们可以保留结晶水。PDB文件里有时会出现其他溶剂或磷酸盐。这些不会影响蛋白质的功能,因此可以将其去除。
E Missing electron density (amino acids)
有时,PDB缺少电子密度,但是,在实际实验中,必须要补全缺失的氨基酸。可以采用以下操作查看蛋白是否完整:
在晶体结构对应的论文中,可以了解结构的全貌,在VMD中看到蛋白质结构。Amber中对参与二硫键的半胱氨酸以CYX代替CYS。
G Protonation States
请记住,我们的最终目标是模拟现实,这可能不一定反映在PDB结构中。其中一个重要的例子是质子化状态。蛋白质含有几种非标准质子化状态的氨基酸。例如,天冬氨酸蛋白酶在其活性位点有一个质子化的天冬氨酸残基。在尝试模拟蛋白质之前,你应该知道你的蛋白质及其功能,你应该了解它是否需要任何非标准的质子化状态。
用X射线方法解析的的晶体结构不含氢,因为该方法无法解析。LEaP根据最佳氢键自动向这些结构中添加氢,同时遵循标准质子化状态。因此,如果不进行必要的重新命名更改,具有非标准质子化状态的氨基酸将被错误质子化。
例如,在天冬氨酸蛋白酶的PDB中,质子化的天冬氨酸将以Asp的重新命名,就像所有其他羧酸天冬氨酸一样。这将导致LEaP质子化,就像它是一种普通的天冬氨酸一样。为了防止这种情况,必须将非标准Asp的别名更改为ASH(质子化Asp的昵称)。使用正确的别名,LEaP将正确质子化您的氨基酸。表1显示了一些常见质子化状态的别名
Table 1. AMBER Resnames of Common Non-standard Protonation States
蛋白结构本身没有问题后,可以进行后续的操作。
小分子结构准备
准备好的小分子还在蛋白结构中,我们对其小分子进行准备,通过以下命令生成化合物的拓扑文件。
antechamber -i lig1.mol2 -fi mol2 -o lig.mol2 -fo mol2 -c bcc -s 2
其中antechamber是调用的程序,-i是输入,-f是格式,-o是输出,-c bcc是采用AM1-BCC电荷模型计算原子点电荷,-s 2是提供verbosity状态信息
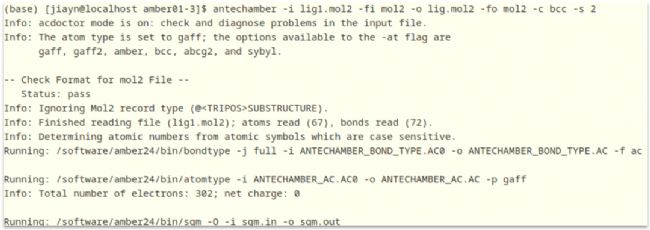
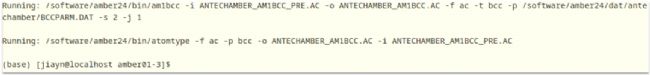
parmchk2 -i lig.mol2 -f mol2 -o lig.frcmod
其中lig.frcmod是一个参数文件,可以加载到LEaP中以添加缺失的参数。这里它将包含所有缺失的参数。
tleap -f tleap_lig.in
对应的脚本可以从官网,或者私信获得。
复合物结构的准备
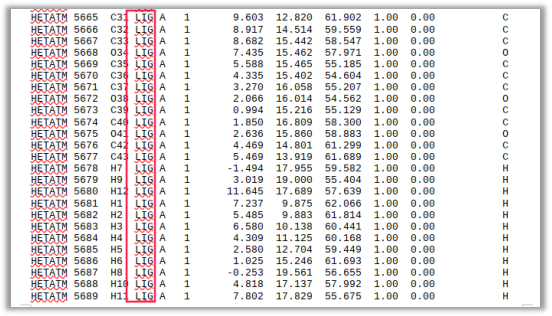
将复合物结构中化合物的编号改为LIG。
输入如下命令,添加离子中和体系电荷、加入水和0.15M的NaCl
tleap -f tleap_complex.in #隐式溶剂模型
tleap -f tleap_ion_water.in #显式溶剂模型
需要计算电荷,可以参考脚本。
体系弛豫
输入命令:
bash all_relax.scr
注意,对应的 min.in 脚本提前准备好,可以从官网下载也可以,另外脚本如果没有执行权限,记得用 chmod 加上权限。对应的解释可以在官网或者私信。
运行动力学
输入命令:
bash jobfile_production.sh
对应的md.in脚本可以从官网获取,也可私信。
目前,已经做好全部的动力学模拟实验,后续等待结果,最后。可以做一些分析,如下:
接下来,让我们一起回顾一下之前的实验结果,并探讨后续推荐的实验思路。
结果分析思路
Apiopaeonoside 反向找靶的结果如下图所示:
假设本次实验发现 Apiopaeonoside 能治疗慢性阻塞性肺疾病、高血压,而预测的第一个靶点 Epoxide hydrolase 刚好和这两种疾病相关,所以,第一个靶点虽然打分(Probability)较低,但是,其仍然值得我们尝试后续的实验。
Sciadopitysin反向找靶结果如下图所示:
假设 Sciadopitysin 有治疗卵巢癌的效果,那么 Poly [ADP-ribose] polymerase 2 打分虽然不如 Poly [ADP-ribose] polymerase-1 好,但是,Poly [ADP-ribose] polymerase 2 和卵巢癌息息相关,所以,Poly [ADP-ribose] polymerase 2 仍然是我们首先考虑的靶点。
后续实验验证
根据假设,目前已经得到了两个分子可能的靶点。后续一般可以用 分子亲合力实验 来证明小分子和蛋白确实是有结合的,比如SPR(表面等离子体共振技术)、MST(微量热泳动技术)。实验需要用到纯化蛋白和小分子单体及对应的仪器,蛋白和小分子陶术生物都可以提供,如果没有实验仪器,也可以找陶术生物来做验证实验!
除上述说的湿实验验证方法外,也可以尝试采用动力学作为补充实验。如黄以超团队在“Environmentally realistic dose of tire-derived metabolite 6PPD-Q exposure causes intestinal jejunum and ileum damage in mice via cannabinoid receptor-activated inflammation ”中所做实验一样。
分子动力学模拟
本次实验将以 Sciadopitysin 和 Poly [ADP-ribose] polymerase 2 复合物结构为演示,采用 Amber24/Ambertool24 进行分子动力学模拟实验。本次实验细节内容将在陶术小课堂中展示。内容较多,会分步介绍,欢迎扫码关注~
首先,我们对得到的结构进行调整,我们需要注意以下7点:
A Non-Standard Residues
B Metals
C Experimental methods noted in the paper associated with the PDB
D Solvent molecules or crystallization buffer
E Missing electron density (amino acids)
F Disulfide Bonds
G Protonation States
A Non-Standard Residues
Non-Standard Residues(非标准残基)是指PDB中20种天然氨基酸以外的其它氨基酸或者分子,包括辅因子(NADH、血红素等)、非标准氨基酸(羟脯氨酸等)、抑制剂/激动剂或底物。如果有任何非标准氨基酸,我们应该考虑如何对这些残基进行建模。对于不含金属或类金属的小有机分子,构建其结构和使用antechamber一样简单。对于更复杂的非标准残基,需要使用别的方法。
B Metals
金属的处理比较复杂,如果复合物结构中包含金属,应该仔细考虑如何对其建模。可以参考amber软件手册。或者在以下两个位置查看软件目前已有的金属力场$AMBERHOME/dat/leap/lib 和 $AMBERHOME/dat/leap/parm。
C Experimental methods noted in the paper associated with the PDB
使用PDB晶体结构的时候,一定要好好阅读一下对应的文献,并注意任何重要的结构特征(比如二硫键)。还要注意用于解析结构的实验程序。有任何问题,都可以直接编辑PDB文件来修复。注意以下几点:
- 第一列中的HEATM标签意味着这些残基默认情况下不会通过键与周围的氨基酸连接。查找/替换HEATM为“ATOM”,使序列连续。请确保保留ATOM后面的两个空格,以便其余列将排列整齐。
- 第二列是原子序号,不要修改
- 第三列是原子的名称。例如,CA是α-碳。因为蛋氨酸不含硒,如果存在,我们需要将这个原子变成硫。将原子名称从SE编辑为SD(蛋氨酸中硫原子的原子名称)。将最后一列中的SE也更改为S。
- 第四列是上面提到的残基名称。将所有MSE条目更改为MET。
在某些情况下,溶剂或结晶缓冲液可能与蛋白质一起结晶,并包含在PDB文件中。因为我们后续采用显式溶剂体系,所以我们可以保留结晶水。PDB文件里有时会出现其他溶剂或磷酸盐。这些不会影响蛋白质的功能,因此可以将其去除。
E Missing electron density (amino acids)
有时,PDB缺少电子密度,但是,在实际实验中,必须要补全缺失的氨基酸。可以采用以下操作查看蛋白是否完整:
- 使用VMD显示蛋白质骨架,并寻找任何缺失。
- 或者,您可以在PDB文件中查找缺失的残基。
在晶体结构对应的论文中,可以了解结构的全貌,在VMD中看到蛋白质结构。Amber中对参与二硫键的半胱氨酸以CYX代替CYS。
G Protonation States
请记住,我们的最终目标是模拟现实,这可能不一定反映在PDB结构中。其中一个重要的例子是质子化状态。蛋白质含有几种非标准质子化状态的氨基酸。例如,天冬氨酸蛋白酶在其活性位点有一个质子化的天冬氨酸残基。在尝试模拟蛋白质之前,你应该知道你的蛋白质及其功能,你应该了解它是否需要任何非标准的质子化状态。
用X射线方法解析的的晶体结构不含氢,因为该方法无法解析。LEaP根据最佳氢键自动向这些结构中添加氢,同时遵循标准质子化状态。因此,如果不进行必要的重新命名更改,具有非标准质子化状态的氨基酸将被错误质子化。
例如,在天冬氨酸蛋白酶的PDB中,质子化的天冬氨酸将以Asp的重新命名,就像所有其他羧酸天冬氨酸一样。这将导致LEaP质子化,就像它是一种普通的天冬氨酸一样。为了防止这种情况,必须将非标准Asp的别名更改为ASH(质子化Asp的昵称)。使用正确的别名,LEaP将正确质子化您的氨基酸。表1显示了一些常见质子化状态的别名
| Non-standard Protonation Form | AMBER Resname |
| Protonated/uncharged Asp | ASH |
| Protonated/uncharged Glu | GLH |
| Deprotonated/uncharged Lys | LYN |
| His protonated at epsilon position | HIE |
| His protonated at delta position | HID |
| Charged His (protonated at both positions) | HIP |
| Deprotonated Cys or Cys bound to a metal | CYM |
| Cys involved in disulfide bridge | CYX |
蛋白结构本身没有问题后,可以进行后续的操作。
小分子结构准备
准备好的小分子还在蛋白结构中,我们对其小分子进行准备,通过以下命令生成化合物的拓扑文件。
antechamber -i lig1.mol2 -fi mol2 -o lig.mol2 -fo mol2 -c bcc -s 2
其中antechamber是调用的程序,-i是输入,-f是格式,-o是输出,-c bcc是采用AM1-BCC电荷模型计算原子点电荷,-s 2是提供verbosity状态信息
▲输出之后对应的界面
▲完成的界面
parmchk2 -i lig.mol2 -f mol2 -o lig.frcmod
其中lig.frcmod是一个参数文件,可以加载到LEaP中以添加缺失的参数。这里它将包含所有缺失的参数。
tleap -f tleap_lig.in
对应的脚本可以从官网,或者私信获得。
复合物结构的准备
将复合物结构中化合物的编号改为LIG。
▲复合物结构的修改
输入如下命令,添加离子中和体系电荷、加入水和0.15M的NaCl
tleap -f tleap_complex.in #隐式溶剂模型
tleap -f tleap_ion_water.in #显式溶剂模型
需要计算电荷,可以参考脚本。
体系弛豫
输入命令:
bash all_relax.scr
注意,对应的 min.in 脚本提前准备好,可以从官网下载也可以,另外脚本如果没有执行权限,记得用 chmod 加上权限。对应的解释可以在官网或者私信。
运行动力学
输入命令:
bash jobfile_production.sh
对应的md.in脚本可以从官网获取,也可私信。
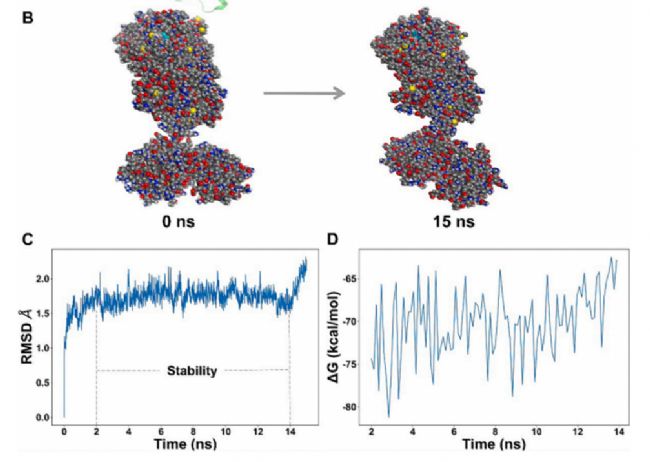
目前,已经做好全部的动力学模拟实验,后续等待结果,最后。可以做一些分析,如下:
动力学结果样图
相关文章
更多 >