

GWAS全基因组关联分析第一期:数据及格式转换
2025-06-12 来源:本站 点击次数:1798GWAS(全基因组关联研究,Genome-Wide Association Study)是一种通过扫描全基因组范围内的遗传变异(如单核苷酸多态性,SNP),寻找与特定性状或疾病显著关联的基因位点的方法。
在进行GWAS分析之前,需要选择合适的群体,进行数据收集。实际工作中发现有些客户根本不知道GWAS分析需要什么数据,耽误分析进度,那需要收集哪些数据呢?主要需要两个数据:表型数据和基因型数据。
01 表型数据
谈起表型数据,我们先来了解下GWAS的表型性状。GWAS中的表型性状可以分为以下三类:
① 数量性状(Quantitative Traits):是指可以用数字值来描述的性状。比如身高(cm)/体重(kg)/籽粒数(个)/产量(kg/亩),分析时表型直接使用具体数值;
② 质量性状(Qualitative Traits):与数量性状相反,其无法用固定数值表示,而是表现出一种状态。比如花色(红、黄、白等)/果实形状(圆、椭圆等)/疾病(有或无),表型可以用数值简化表示;
③ 分级性状(Ordinal Traits):是介于质量性状和数量性状之间的一类性状,表现为有序的类别,但这些类别之间的差异不是连续的。比如病毒抵抗性水平(高、中、低)/籽粒颜色(浅黄、深黄、棕色等)/植株高度级别(高、中、低),这些性状可以用数字值(1、2、3等)来描述。
那表型文件长啥样呢?这里以常见疾病研究为例,文件一般包含3列:样本(Sample)、性别(Sex)和表型(Phenotype)。
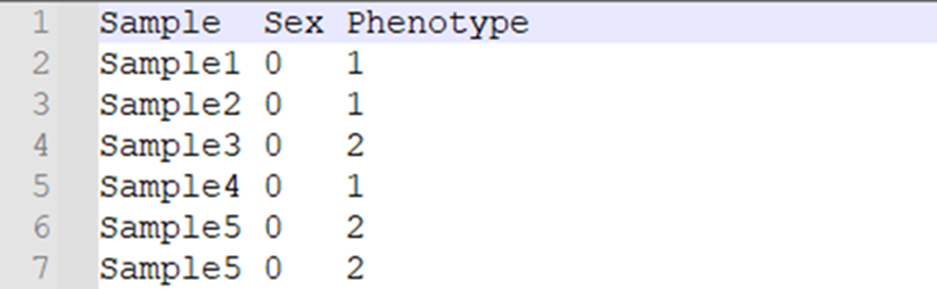
注意:性别用数值表示,0表示未知、1表示男、2表示女、-9表示缺失;表型也用数值表示,0表示未知、1表示对照组、2表示实验组、-9表示缺失。不要出现空缺值!
02 基因型数据
基因型是指一个个体在某个特定基因位点上所拥有的等位基因的组合。每个基因位点可以有不同的等位基因,这些等位基因是由父母各自传递的一个单倍体组成。
那基因型数据从哪里获取呢?通常有两种方式可以获取:
① 基于SNP芯片获取基因型数据
② 基于基因组测序获取基因型数据
这里我们以VCF文件为例,从”#CHROM”列开始就是变异数据:
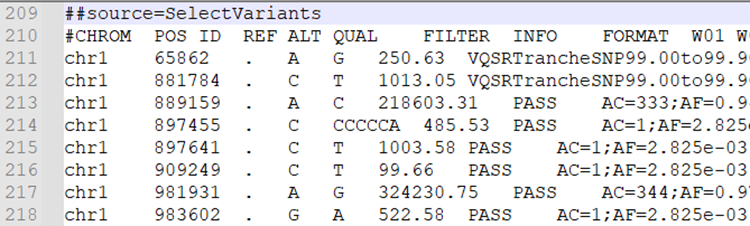
注意:由于原始变异文件中有大量不可靠变异,建议使用过滤后的VCF文件(比如筛选PASS)进行后续分析。
由于GWAS分析需要动辄几百上千例的数据,客户很难凑齐那么多样本,特别还要额外的正常样本作为对照,对客户而言又是一笔很大的支出!这个时候我们可以采用既有的正常人数据作为对照组,最典型的就是使用千人基因组计划数据为对照。
现在表型数据和基因型数据都有了,该如何使用呢?由于整个关联分析流程是基于PLINK软件,那么就需要将基因型数据转换为PLINK软件能识别的格式,同时也利于提高数据处理效率。PLINK中有两类数据格式,在分析过程都会用到:
1. ped/map格式
> ped格式文件:
包含每个样本的基本信息(族系、名称、性别等)、表现型信息(phenotype)、基因型信息(第7列之后)。每行表示一个样本,该文件没有表头,具体列数取决于该样本所含SNPs位点数。
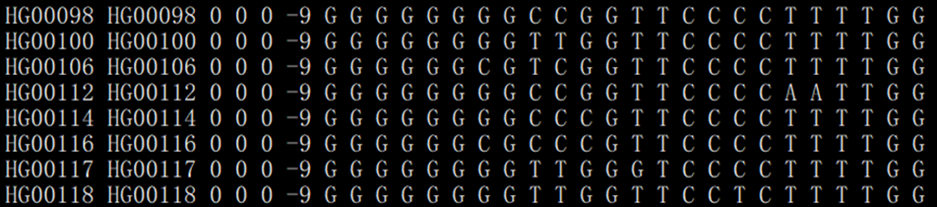
第一列:Family ID,族系ID;
第二列:Individual ID,个体(如样本间无族系联系,Family ID和Individual ID可以一样);
第三列:Paternal ID,父系ID,0表示未知,-1表示无父亲/缺失;
第四列:Maternal ID,母系ID,0表示未知,-1表示无母亲/缺失;
第五列:Sex,性别,1 男,2 女,0 或 其他值 表示未知;
第六列:Phenotype,表型值,根据研究类型定义;
后续列:从第七列开始,每两列代表该样本所含的一个SNP的基因型:如第七、八列代表第一个基因型(GG),第九、十列代表第二个基因型(GG)等等。
> map格式文件:
记录每个 SNP 的染色体位置和遗传学信息。每行对应一个 SNP,该文件没有表头,每行包含四列。
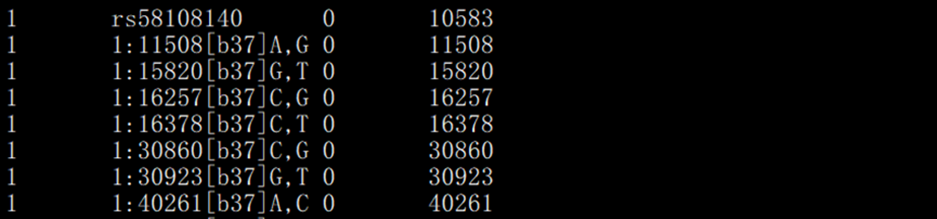
第一列:染色体编号;
第二列:变异标识符,这里是rs编号(有的话)或其他方式;
第三列:遗传距离(摩尔根,单位cM),未知情况下写0即可;
第四列(可选):SNP在染色体上物理位置,跟第三列必有一列。
2. bed/bim/fam格式
> bed格式文件:
存储基因型信息(二进制),每行对应一个样本。由于是二进制格式不能直接打开。
第一列:样本的 ID(需与 .fam 文件中的 IID 一致);
后续列:每两列为一个 SNP 的两个等位基因(按顺序排列,如 A/C 或 0/1)。其中0、1、2分别对应了aa、Aa或aA和AA。
> bin格式文件:
存储每个 SNP 的元信息(染色体位置、遗传学距离等)。每行对应一个SNP。
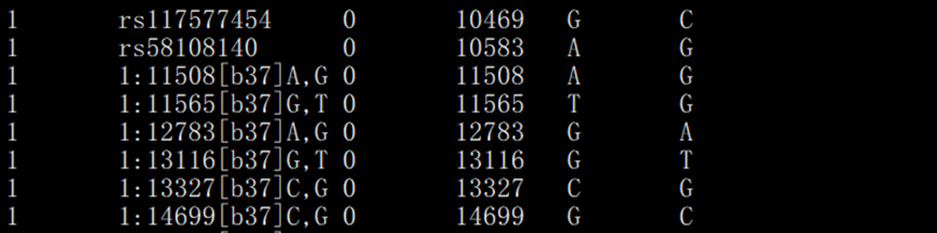
第一列:Chr,染色体编号;
第二列:SNP,标记名称;
第三列:GD,遗传距离(摩尔根),未知情况下写0;
第四列:BPP,变异位点物理位置(单位:bp);
第五列:Allele 1,一般情况下为次要等位基因;
第六列:Allele 2,一般情况下为主要等位基因。
> fam格式文件:
存储样本的家系关系和表型信息,每行对应一个样本。

第一列:FID,家系ID;
第二列:IID,个体ID;
第三列:父本个体ID,没有用0表示,-1表示缺失;
第四列:母本个体ID,没有用0表示,-1表示缺失;
第五列:SEX,性别,1表示男性,2表示女性,0表示未知;
第六列:Phenotype,表型值。
小结:fam文件就是ped文件前六列。此外这里可以通过手动添加补全表型、性别信息。
那怎么通过vcf格式数据得到ped/map格式和bed/bim/fam格式数据呢?直接上命令:
vcf转ped/map格式:
plink --vcf test.vcf.gz --recode --out test
vcf转bed/bim/fam格式:
plink --vcf test.vcf.gz --make-bed --out test
ped/map转bed/bim/fam格式:
plink --file test --make-bed --out test
bed/bim/fam转ped/map格式:
plink --bfile test --recode --out test
以上就是本期分享的内容,下一期我们将讲解如何对plink格式数据进行质控过滤。