

现代制药行业中计算机化系统验证的执行
2025-11-20 来源:本站 点击次数:1137生物制药行业中,计算机化系统正在占据越来越大的比重,相较于依靠记录人员熟练度和专注性的人工记录,电子记录对ALCOA+原则的执行性对于产品的风险分析及稳定放行是至关重要的,这不仅是为了保证产品的质量,更是为了保障患者的安全。为了确保数字化工具可靠、合规、数据完整,对应的验证工作必不可少,其中FDA/EU/NMPA均对计算机化系统的功能,记录,验证有明确详细的要求。
计算机化系统验证(CSV)的执行规则并非一成不变,在针对不同的流程,产品以及工具属性的情况下,更倾向于“法律条文”、“行业标准”和“内部章程” 的结合,核心为ISPE的GAMP 5,要求在工艺开发及生产的流程中,基于风险,效率,合规对执行和记录的控制。在一切开始之前,应当对不同系统的风险等级进行评估,一个用于计算标准产量的表格,与一个用于放行药品的质量标准,其验证强度和深度等级完全不同,应当将主要的工作内容聚焦于关键设备及关键功能。在验证路径中,GAMP 5建立了一个“V模型”用于展示每一个设计与测试之间的追溯关系,即每一个测试都应当可追溯至对应的工艺需求或合规设计,每一个执行的标准都应当有对应的证据链条。
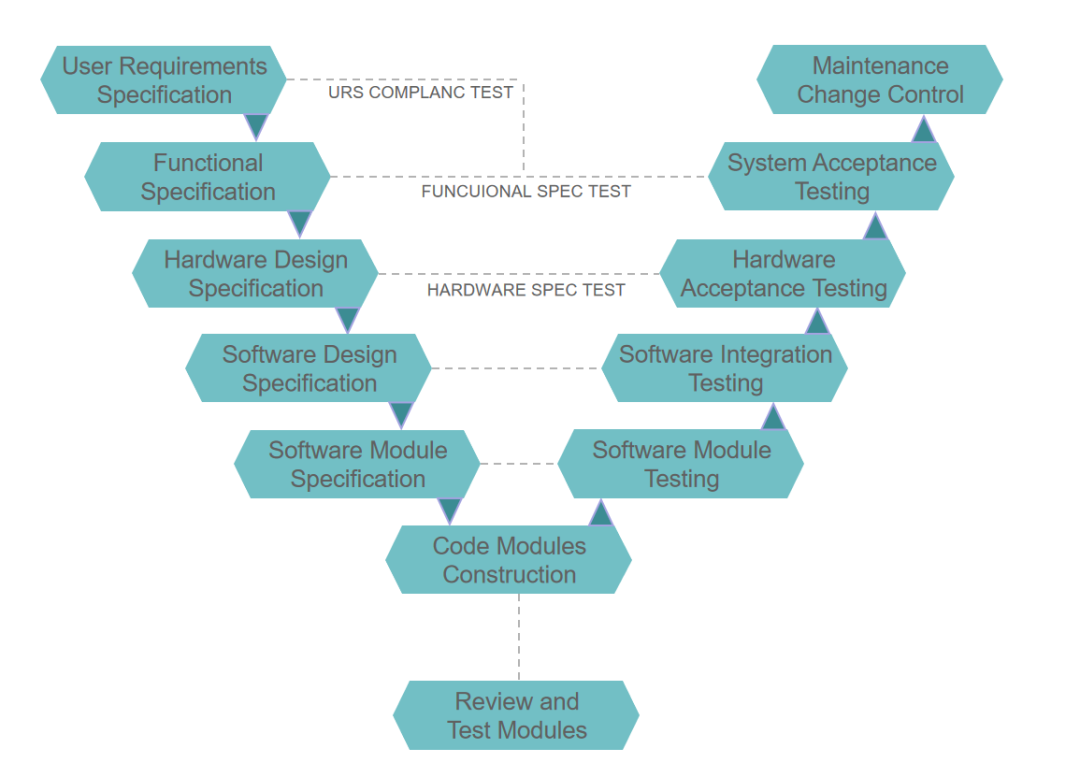
V模型
整体数据记录的来源及内容,其来源均对应于法规的监管内容。对于整个生命周期内的保障,应参考EU GMP 附录 11中对风险评估、供应商管理、数据完整性、变更管理、定期回顾、退役管理的相关要求,其正是对GAMP 5理念在监管方面的执行体现。同时为了保证电子记录及签名对纸质记录的实质可替代性,应依据FDA 21 CFR Part 11中的核心要求,在一切开始之前,确定人员培训、系统验证、审计追踪、电子签名绑定、权限控制、记录保护等对应测试内容。以《GAMP 5》为方法论,以《CFR Part 11》、《Annex 11》等法规为底线,以《公司质量体系文件》为内部执行标准,以《变更控制》和《定期回顾》为持续保障,才能最终获得完整可信的电子数据。
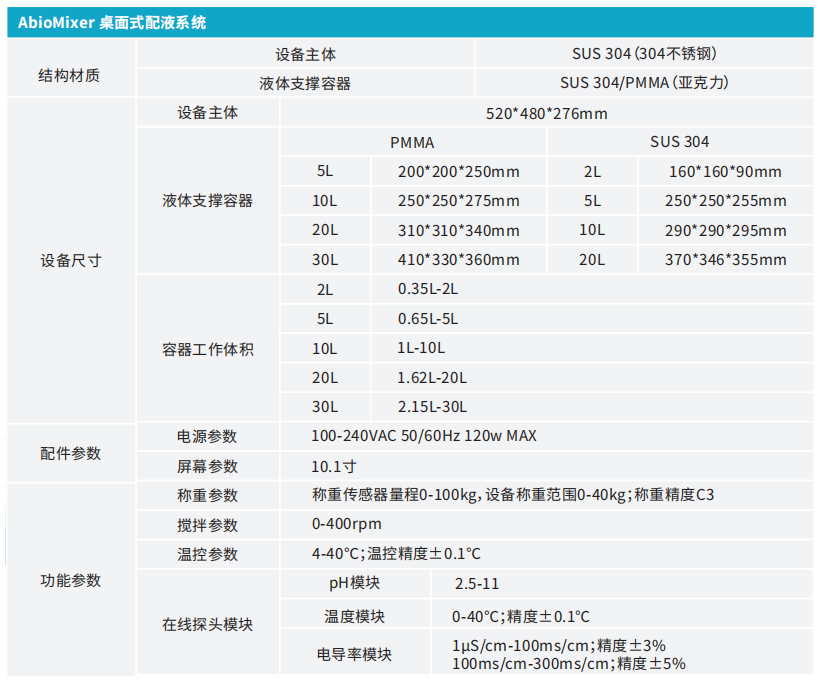
AbioMixer桌面式配液系统艾贝泰一次性工艺配液系统,从设计到生产全流程贴合当下愈加激烈的竞争态势和审查要求,从而符合CSV的严格标准。

新成员AbioMixer®桌面式配液系统基于角色的动态权限控制,内嵌的审计追踪模块完整真实的记录您的每一次操作,从源头杜绝人工转录错误,保障数据ALCOA+原则,与您的质量体系完美契合,是为生物制药量身打造的合规引擎。从严格的电子化信息记录,到精细化的权限矩阵管理,可靠的上位机连接方案与数据自动采集功能,为您的项目全流程保驾护航,一站式软硬件设计流程,免除繁杂的物料及方案设计,协助项目快速落地。
同时,基于服务器功能的配方管理及控制,在满足常规单机设备自动化要求中对单个步骤的速率及时间控制基础上,额外增加了自动/手动判断跳转,自动提示,步骤确认,外置功能模块定制化等功能,为满足不同项目阶段,不同工艺流程的需求,降低您的使用成本。
应用场景:
商业化生产流程中针对缓冲液及中间产品的小体积混匀需求。
项目研发小试阶段的培养基、缓冲液准备及产品混匀需求。
中试阶段工艺放大中,混匀工艺参数的来源。
应用领域:
细胞和基因治疗:用于培养基、缓冲液配置,以及病毒转染前的多质粒混匀等。
疫苗制剂:用于多价疫苗制剂前的混匀,以及中间产物和半成品的配制等。
生物制药:用于各种生物制药的工艺开发和生产,包括蛋白质工程、细胞培养和药物筛选等。
制药流程控制的可信度不是一种感觉,而是由每一份文件的签字确认、每一条测试的通过标记、每一套管理程序的履行记录所共同构筑的、客观存在的质量保证。通过需求规范-设计规范-风险评估-验证确认,获得一份完整、连贯、可审计的证据链,证明系统在整个生命周期内,持续地、可预测地满足其既定用途,并符合法规要求,是产品可信度的证明,也是企业长期平稳运行的保障。