

利用高温酸性酶解确定未知翻译后修饰并提高蛋白质序列覆盖率
2026-04-28 来源:本站 点击次数:464
在蛋白质组学搜库分析中,设置合适的翻译后修饰 (PTM) 对于尽可能全面地获取数据结果至关重要。但一般在搜库前,并不能确定应选择哪些 PTM,尤其是在非典型酶解条件下(如使用超嗜热酸性古菌 (HTA) 酶进行酶解时采用的高温、低 pH 条件),样品制备过程中产生的PTM可能与常规胰蛋白酶酶解产生的PTM存在较大差异。
本研究采用 PEAKS® Studio 软件中的 PEAKS PTM 功能,对比分析胰蛋白酶酶解产物与HTA酶解产物中的PTM差异。结果显示,HTA蛋白酶数据的蛋白质鉴定数量增加了 8%,且仅通过HTA酶解鉴定出的蛋白质数量从 91 个提升至 248 个。
前言
蛋白质组学理想状态是全面鉴定样品中所有蛋白质并实现其翻译后修饰 (PTM) 的全覆盖,但现有方法仅能鉴定部分蛋白及修饰。通常 bottom-up 流程采用胰蛋白酶在碱性条件下酶解,酶的特异性高,对富含赖氨酸和精氨酸残基的蛋白质鉴定更加友好;同时样品制备过程中(如高温、变性剂处理等)也可能产生非酶促PTM(如氧化、脱酰胺等),这些也通常被纳入组学数据的分析中。
Krakatoa 酶和 Vesuvius 酶是超嗜热酸性古菌 (HTA) 酶的典型代表。在高温(约 80℃)、低pH(约 3)条件下活性最佳,在样品制备过程中无需添加变性剂或烷基化处理,其半特异性酶切特性可鉴定胰蛋白酶酶解所遗漏的蛋白序列与蛋白质,且酶解速度极快。但HTA酶与胰蛋白酶反应条件差异大,常规胰蛋白酶搜索中使用的PTM并不适用于HTA酶解数据。大多数组学软件都要求用户在搜库开始前先定义PTM,但设置大量可变PTM会导致搜索空间扩大和假阳性的问题,影响分析效率与效果。
PEAKS® Studio 软件中的 PEAKS PTM 算法可解决上述问题,支持无限制的PTM搜索。其工作流首先对所有二级质谱 (MS2) 谱图进行从头测序,然后进行数据库搜索。对于从头测序得分较高但未与数据库匹配的谱图,无需预先指定PTM,只需一键勾选 "PEAKS PTM" 即可自动搜索[2]。这种搜索方法可有效避免常规数据库搜索策略中大量可变修饰导致的搜索空间大且假阳性高的问题。
本研究应用 PEAKS PTM 算法,搜索并对比分析了胰蛋白酶与HTA酶分别酶解K562细胞蛋白的PTM及蛋白质鉴定结果。 HTA酶解数据除设置常规PTM之外,加入额外的PTM搜索可提高蛋白质覆盖率,同时还鉴定出胰蛋白酶酶解无法识别的蛋白质。
研究方法
我们使用 PEAKS® Studio (BSI) 对公开数据集 PXD041226[1]进行重新分析。该数据集包含K562细胞裂解液经胰蛋白酶、Krakatoa 酶、Vesuvius 酶分别酶解的样品,每种酶解条件设置三个重复,共 9 个样品。所有样品均采用 Thermo Fisher Scientific 公司的 Fusion Lumos Tribrid 质谱仪进行分析。
在原始文献中,所有数据文件通过 PEAKS® Studio XPro 进行联合搜索,酶切位点设置为非特异性,并设置可变修饰(详见参考文献)。
使用 PEAKS® Studio 13 对数据进行重新分析,参数如表 1 所示。分别在勾选和不勾选 PEAKS PTM 算法的情况下进行数据搜索,并根据所用酶的不同,将数据分为三组进行单独搜索。
研究结果
表 2 展示了通过 PEAKS PTM 搜索得到的每种酶对应的 Top 10 PTM(按鉴定出的 unique 位点数量排序)。从列表中可见,脱酰胺作用 (NQ)、蛋白质N端乙酰化和钠加合物是胰蛋白酶与HTA酶共有的PTM。然而,采用这种方法难以对比不同酶之间每种PTM的修饰程度。这不仅因为每种酶搜索得到的肽段数量不同,还因为鉴定到的肽段因氨基酸序列不同,导致其发生特定PTM的潜在可能性存在差异。
为解决这一问题,我们用 “已鉴定的PTM位点总数” 与 “该PTM所有可能的 unique 修饰位点总数”, 计算出每种PTM的可能的修饰位点百分比。这种“标准化”处理使不同样品之间具有了可比性,从而能够对比不同酶之间的PTM修饰程度。
表 3 展示了通过 PEAKS PTM 搜索得到的每种酶对应的 Top 10 PTM(按可能位点修饰百分比排序); Deamidation, N-terminal acetylation, oxidation 和 sodium adduct 在胰蛋白酶与HTA酶的PTM鉴定结果中均排名靠前。与预期不同的是, oxidation 和 Deamidation 在HTA酶解产物中的排名也很高,因为这些修饰通常被认为与胰蛋白酶酶解的碱性条件更相关。
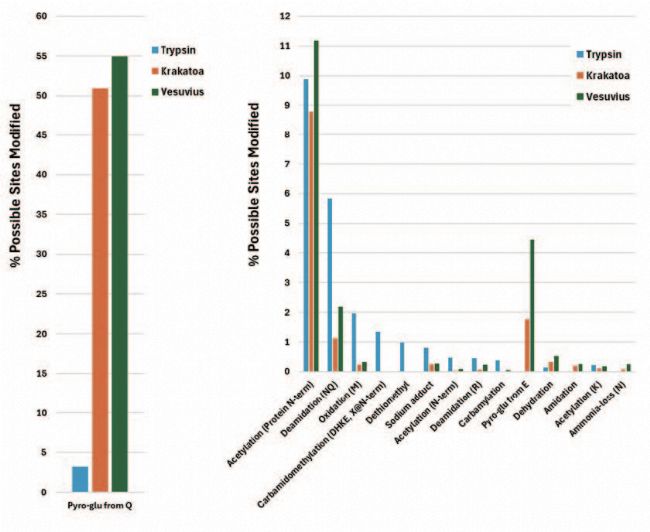
Figure 1. Bar graphs depicting the percentage of sites modified with various PTMs out of all sites that could be possibly modified for different enzymes. Pyro-glu from Q is in a separate graph due to the different scale compared to other PTMs.
由于胰蛋白酶酶解发生在碱性条件下,半胱氨酸通常需进行烷基化处理以防止二硫键形成。碱性条件还可能加剧脱酰胺与氧化修饰的发生,其中甲硫氨酸氧化尤为常见,会进一步干扰后续质谱鉴定与序列分析。此外,蛋白变性过程中使用尿素也会增加氨甲酰化的发生概率。相反,在HTA酶解产物中, pyro-glu from Q or E 是修饰位点占比最高的PTM,超过 50% 的N端谷氨酰胺潜在位点发生了这种修饰,远高于胰蛋白酶酶解产物。高温条件可能是焦谷氨酸形成的常见原因,这也解释了为何HTA酶解产物中该PTM含量极高。
此外,HTA酶与胰蛋白酶的酶切位点不同,可能会产生更多N端含谷氨酰胺的肽段,进而增加了这类PTM被检测的概率。脱水作用和酰胺化在HTA酶解产物中更为常见,但与焦谷氨酸相比,其修饰位点占比仍较低(不足 1%),这可能是因为这些PTM在酸性条件下更易形成。
蛋白质N端乙酰化作为一种常见的体内生物学PTM,在不同酶解产物中的水平相对一致。肽段末端产生修饰可能发生在酶解过程中或酶解后,且可能与酶解条件相关。胰蛋白酶酶解产物中肽段N端乙酰化更为常见,说明胰蛋白酶酶解条件更有利于乙酰化反应的发生。
综合PTM鉴定位点数量和潜在位点修饰百分比两方面因素,HTA酶解数据分析中应优先考虑纳入的 Top PTM 包括: deamidation (NQ), acetylation (protein N-term), pyro-glu from Q, dehydration, pyro-glu from E, amidation 和 sodium adduct。若采用传统数据库搜索方法,非必要情况下,不建议设置 dehydration 和 sodium adduct 等容易发生在多个氨基酸上的修饰。但使用 PEAKS PTM 算法则无限制,可设置所有感兴趣的PTM。
我们还用 PEAKS® Studio 13 对 9 个样品的进行了联合搜索,用于展示 PTM Profiling 工具的功能。在搜索过程中,可变修饰设置 N-terminal protein acetylation, carbamidomethylation (at cysteine), deamidation 和 pyro-glu form Q(每个肽段最多允许 3 个修饰),同时勾选 PEAKS PTM 功能。
PEAKS® Studio 13 的 PTM Profiling 工具使用包含特定修饰位点的肽段的一级质谱 (MS1) 特征峰面积,计算该位点的修饰比例和未修饰比例。
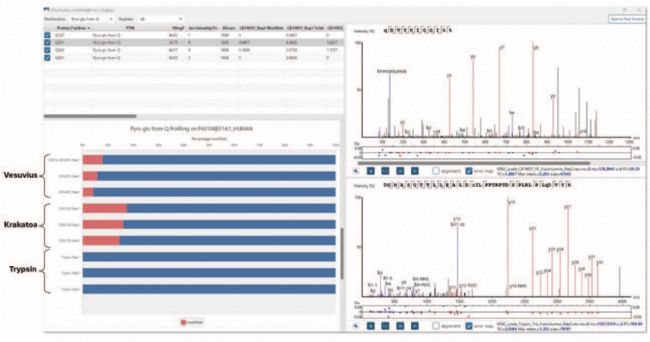
Figure 2. The PEAKS Studio 13 PTM profiling tool. Pyroglutamate formation at site Q251 on the protein Elongation Factor 1-alpha is shown.
将 PEAKS PTM 搜索结果与原始文献结果对比发现(图 3),两种HTA酶解产物的搜索结果均覆盖了原始文献中超过 90% 的肽段和 85% 的蛋白质,且 PEAKS PTM 算法还鉴定出了原始分析中未发现的部分肽段和蛋白质。这是因为本篇研究根据酶解所用酶的不同对样品进行分组搜索,未纳入无关PTM,大幅提升了搜索效率;PEAKS® Studio 13 的搜索算法整合了深度学习的重打分功能,相比 PEAKS® Studio XPro 显著提升分析结果质量;纳入原始分析中未考虑的PTM也可能对结果提升起到了一定作用(图 4-6)。
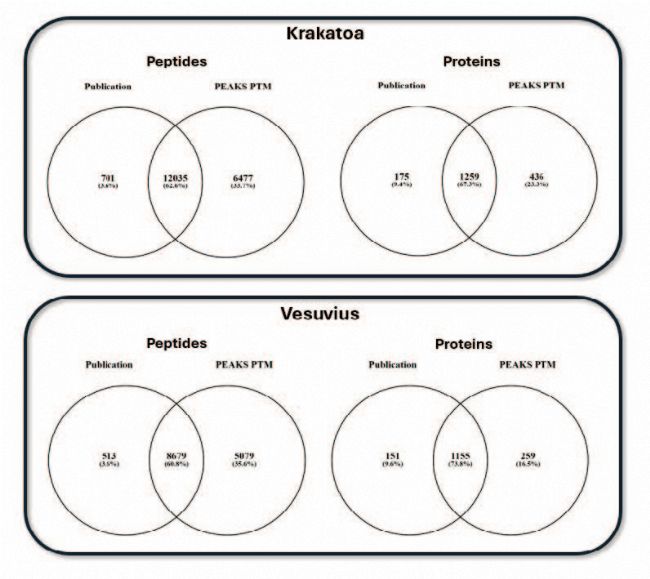
Figure 3. Comparison of results from original publication to reanalysis with PEAKS PTM. The new search of the Krakatoa digest covers 94% of the original peptide identifications, with 6477 additional peptides and 88% of the original protein identifications, with 1434 additional proteins. The new search of the Vesuvius digest covers 94% of the original peptide identifications, with 9192 additional peptides and 89% of the original protein identifications, with 1306 additional proteins.

Figure 4. Krakatoa coverage outline of Elongation Factor 1-alpha using a search without PTMs and with PEAKS PTM. The individual peptide coverage and redundancy of the first 80 amino acids is shown.

Figure 5. Vesuvius coverage outline of Elongation Factor 1-alpha using a search without PTMs and with PEAKS PTM. The individual peptide coverage and redundancy of the first 80 amino acids is shown.
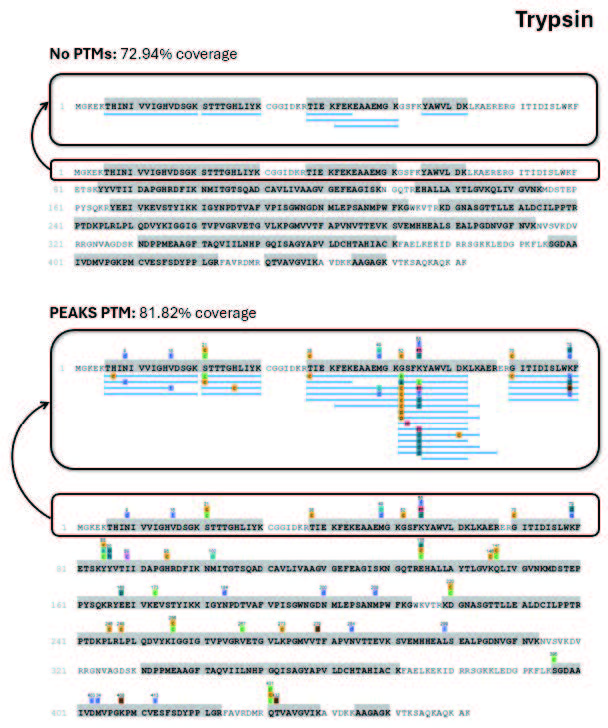
Figure 6. Trypsin coverage outline of Elongation Factor 1-alpha using a search without PTMs and with PEAKS PTM. The individual peptide coverage and redundancy of the first 80 amino acids is shown.
此外,我们对比了不设置任何可变修饰与运行 PEAKS PTM 算法两种情况下的搜索结果(参数如表 1 所示),并以在所有鉴定结果中均以可信度较高的 Elon-gation Factor 1-alpha 蛋白为例,设置了PTM后,蛋白质序列覆盖率大幅提升。
所有酶解产物的分析在添加PTM之后均有提升,其中 Krakatoa 和 Vesuvius 酶解产物在运行 PEAKS PTM 算法后,序列覆盖率均超过 99%;而胰蛋白酶酶解产物的序列覆盖率仅为 82%。这表明,将半特异性HTA酶与 PEAKS PTM 搜索工具结合使用,可有效提高蛋白质序列覆盖率。
研究结论
PEAKS PTM 算法因无需预先定义PTM类型,是发现非常规胰蛋白酶酶解条件下PTM的理想方法。HTA酶解产物的PTM水平低于胰蛋白酶酶解产物,这一差异大概率源于二者酶解反应条件不同,且HTA酶解过程耗时更短。但在HTA酶解产物中,仍能鉴定出胰蛋白酶酶解产物中不常见的独特PTM(如 pyro-glu form Q)。
针对不同的酶解条件设置最合适的PTM,有助于提高蛋白质序列覆盖率,并最终帮助研究人员获取传统方法可能遗漏的蛋白质信息。
参考文献
1. McCabe M C, Gejji V, Barnebey A, et al. From volcanoes to the bench: Advantages of novel hyperthermoacidic archaeal proteases for proteomics workflows . Journal of Proteomics, 2023, 289: 104992. https://doi.org/10.1016/j.jprot.2023.104992.
2. Xi Han, Lin He, Lei Xin, Baozhen Shan, and Bin Ma Journal of Proteome Research 2011 10 (7), 2930-2936 DOI: 10.1021/pr200153k
3. Dick LW Jr, Kim C, Qiu D, Cheng KC. Determination of the origin of the N-terminal pyro-glutamate variation in monoclonal antibodies using model peptides. Biotechnol Bioeng. 2007 Jun 15;97(3):544-53. doi:10.1002/bit.21260. PMID: 17099914.
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
本研究采用 PEAKS® Studio 软件中的 PEAKS PTM 功能,对比分析胰蛋白酶酶解产物与HTA酶解产物中的PTM差异。结果显示,HTA蛋白酶数据的蛋白质鉴定数量增加了 8%,且仅通过HTA酶解鉴定出的蛋白质数量从 91 个提升至 248 个。
前言
蛋白质组学理想状态是全面鉴定样品中所有蛋白质并实现其翻译后修饰 (PTM) 的全覆盖,但现有方法仅能鉴定部分蛋白及修饰。通常 bottom-up 流程采用胰蛋白酶在碱性条件下酶解,酶的特异性高,对富含赖氨酸和精氨酸残基的蛋白质鉴定更加友好;同时样品制备过程中(如高温、变性剂处理等)也可能产生非酶促PTM(如氧化、脱酰胺等),这些也通常被纳入组学数据的分析中。
Krakatoa 酶和 Vesuvius 酶是超嗜热酸性古菌 (HTA) 酶的典型代表。在高温(约 80℃)、低pH(约 3)条件下活性最佳,在样品制备过程中无需添加变性剂或烷基化处理,其半特异性酶切特性可鉴定胰蛋白酶酶解所遗漏的蛋白序列与蛋白质,且酶解速度极快。但HTA酶与胰蛋白酶反应条件差异大,常规胰蛋白酶搜索中使用的PTM并不适用于HTA酶解数据。大多数组学软件都要求用户在搜库开始前先定义PTM,但设置大量可变PTM会导致搜索空间扩大和假阳性的问题,影响分析效率与效果。
PEAKS® Studio 软件中的 PEAKS PTM 算法可解决上述问题,支持无限制的PTM搜索。其工作流首先对所有二级质谱 (MS2) 谱图进行从头测序,然后进行数据库搜索。对于从头测序得分较高但未与数据库匹配的谱图,无需预先指定PTM,只需一键勾选 "PEAKS PTM" 即可自动搜索[2]。这种搜索方法可有效避免常规数据库搜索策略中大量可变修饰导致的搜索空间大且假阳性高的问题。
本研究应用 PEAKS PTM 算法,搜索并对比分析了胰蛋白酶与HTA酶分别酶解K562细胞蛋白的PTM及蛋白质鉴定结果。 HTA酶解数据除设置常规PTM之外,加入额外的PTM搜索可提高蛋白质覆盖率,同时还鉴定出胰蛋白酶酶解无法识别的蛋白质。
研究方法
我们使用 PEAKS® Studio (BSI) 对公开数据集 PXD041226[1]进行重新分析。该数据集包含K562细胞裂解液经胰蛋白酶、Krakatoa 酶、Vesuvius 酶分别酶解的样品,每种酶解条件设置三个重复,共 9 个样品。所有样品均采用 Thermo Fisher Scientific 公司的 Fusion Lumos Tribrid 质谱仪进行分析。
在原始文献中,所有数据文件通过 PEAKS® Studio XPro 进行联合搜索,酶切位点设置为非特异性,并设置可变修饰(详见参考文献)。
使用 PEAKS® Studio 13 对数据进行重新分析,参数如表 1 所示。分别在勾选和不勾选 PEAKS PTM 算法的情况下进行数据搜索,并根据所用酶的不同,将数据分为三组进行单独搜索。
Table 1. PEAKS Studio 13 Search Parameters
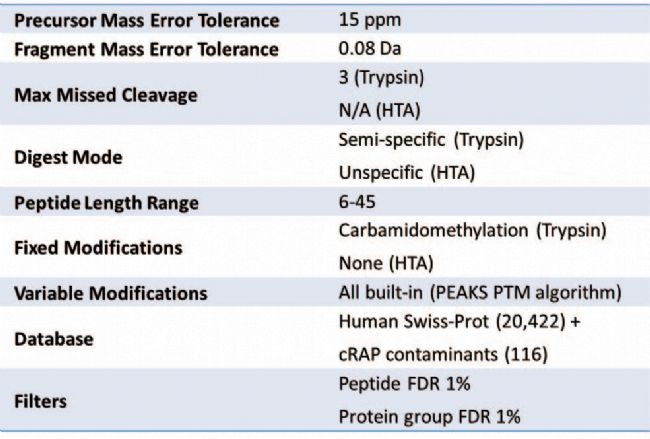
研究结果
表 2 展示了通过 PEAKS PTM 搜索得到的每种酶对应的 Top 10 PTM(按鉴定出的 unique 位点数量排序)。从列表中可见,脱酰胺作用 (NQ)、蛋白质N端乙酰化和钠加合物是胰蛋白酶与HTA酶共有的PTM。然而,采用这种方法难以对比不同酶之间每种PTM的修饰程度。这不仅因为每种酶搜索得到的肽段数量不同,还因为鉴定到的肽段因氨基酸序列不同,导致其发生特定PTM的潜在可能性存在差异。
Table 2. Top 10 PTMs found by PEAKS PTM based on number of unique sites identified.
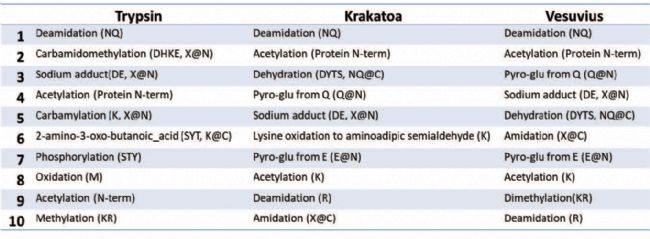
为解决这一问题,我们用 “已鉴定的PTM位点总数” 与 “该PTM所有可能的 unique 修饰位点总数”, 计算出每种PTM的可能的修饰位点百分比。这种“标准化”处理使不同样品之间具有了可比性,从而能够对比不同酶之间的PTM修饰程度。
表 3 展示了通过 PEAKS PTM 搜索得到的每种酶对应的 Top 10 PTM(按可能位点修饰百分比排序); Deamidation, N-terminal acetylation, oxidation 和 sodium adduct 在胰蛋白酶与HTA酶的PTM鉴定结果中均排名靠前。与预期不同的是, oxidation 和 Deamidation 在HTA酶解产物中的排名也很高,因为这些修饰通常被认为与胰蛋白酶酶解的碱性条件更相关。
Table 3. Top 10 PTMs found by PEAKS PTM based on percentage of possible sites modified.
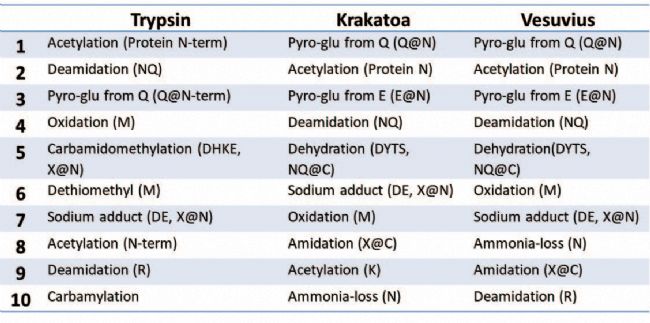
为了直观对比胰蛋白酶与HTA酶的 Top PTM,根据表 3 中的数据绘制柱状图,展示每种PTM的可能修饰位点百分比(图 1)。结果显示,胰蛋白酶在潜在修饰位点上的修饰程度高于HTA酶, Deamidation, oxidation 和 carbamidomethylation 在胰蛋白酶酶解产物中的修饰位点占比均超过 1%,显著高于HTA酶解产物。这表明,尽管这些修饰在HTA酶解产物中也会发生,但总体修饰位点数量相对较少。此外,胰蛋白酶酶解产物中的 Sodium adducts, N-terminal acetylation 和 carbamylation 占比也高于HTA酶解产物,但总体占比均低于其他PTM。
Figure 1. Bar graphs depicting the percentage of sites modified with various PTMs out of all sites that could be possibly modified for different enzymes. Pyro-glu from Q is in a separate graph due to the different scale compared to other PTMs.
由于胰蛋白酶酶解发生在碱性条件下,半胱氨酸通常需进行烷基化处理以防止二硫键形成。碱性条件还可能加剧脱酰胺与氧化修饰的发生,其中甲硫氨酸氧化尤为常见,会进一步干扰后续质谱鉴定与序列分析。此外,蛋白变性过程中使用尿素也会增加氨甲酰化的发生概率。相反,在HTA酶解产物中, pyro-glu from Q or E 是修饰位点占比最高的PTM,超过 50% 的N端谷氨酰胺潜在位点发生了这种修饰,远高于胰蛋白酶酶解产物。高温条件可能是焦谷氨酸形成的常见原因,这也解释了为何HTA酶解产物中该PTM含量极高。
此外,HTA酶与胰蛋白酶的酶切位点不同,可能会产生更多N端含谷氨酰胺的肽段,进而增加了这类PTM被检测的概率。脱水作用和酰胺化在HTA酶解产物中更为常见,但与焦谷氨酸相比,其修饰位点占比仍较低(不足 1%),这可能是因为这些PTM在酸性条件下更易形成。
蛋白质N端乙酰化作为一种常见的体内生物学PTM,在不同酶解产物中的水平相对一致。肽段末端产生修饰可能发生在酶解过程中或酶解后,且可能与酶解条件相关。胰蛋白酶酶解产物中肽段N端乙酰化更为常见,说明胰蛋白酶酶解条件更有利于乙酰化反应的发生。
综合PTM鉴定位点数量和潜在位点修饰百分比两方面因素,HTA酶解数据分析中应优先考虑纳入的 Top PTM 包括: deamidation (NQ), acetylation (protein N-term), pyro-glu from Q, dehydration, pyro-glu from E, amidation 和 sodium adduct。若采用传统数据库搜索方法,非必要情况下,不建议设置 dehydration 和 sodium adduct 等容易发生在多个氨基酸上的修饰。但使用 PEAKS PTM 算法则无限制,可设置所有感兴趣的PTM。
我们还用 PEAKS® Studio 13 对 9 个样品的进行了联合搜索,用于展示 PTM Profiling 工具的功能。在搜索过程中,可变修饰设置 N-terminal protein acetylation, carbamidomethylation (at cysteine), deamidation 和 pyro-glu form Q(每个肽段最多允许 3 个修饰),同时勾选 PEAKS PTM 功能。
PEAKS® Studio 13 的 PTM Profiling 工具使用包含特定修饰位点的肽段的一级质谱 (MS1) 特征峰面积,计算该位点的修饰比例和未修饰比例。
Figure 2. The PEAKS Studio 13 PTM profiling tool. Pyroglutamate formation at site Q251 on the protein Elongation Factor 1-alpha is shown.
将 PEAKS PTM 搜索结果与原始文献结果对比发现(图 3),两种HTA酶解产物的搜索结果均覆盖了原始文献中超过 90% 的肽段和 85% 的蛋白质,且 PEAKS PTM 算法还鉴定出了原始分析中未发现的部分肽段和蛋白质。这是因为本篇研究根据酶解所用酶的不同对样品进行分组搜索,未纳入无关PTM,大幅提升了搜索效率;PEAKS® Studio 13 的搜索算法整合了深度学习的重打分功能,相比 PEAKS® Studio XPro 显著提升分析结果质量;纳入原始分析中未考虑的PTM也可能对结果提升起到了一定作用(图 4-6)。
Figure 3. Comparison of results from original publication to reanalysis with PEAKS PTM. The new search of the Krakatoa digest covers 94% of the original peptide identifications, with 6477 additional peptides and 88% of the original protein identifications, with 1434 additional proteins. The new search of the Vesuvius digest covers 94% of the original peptide identifications, with 9192 additional peptides and 89% of the original protein identifications, with 1306 additional proteins.
Figure 4. Krakatoa coverage outline of Elongation Factor 1-alpha using a search without PTMs and with PEAKS PTM. The individual peptide coverage and redundancy of the first 80 amino acids is shown.
Figure 5. Vesuvius coverage outline of Elongation Factor 1-alpha using a search without PTMs and with PEAKS PTM. The individual peptide coverage and redundancy of the first 80 amino acids is shown.
Figure 6. Trypsin coverage outline of Elongation Factor 1-alpha using a search without PTMs and with PEAKS PTM. The individual peptide coverage and redundancy of the first 80 amino acids is shown.
此外,我们对比了不设置任何可变修饰与运行 PEAKS PTM 算法两种情况下的搜索结果(参数如表 1 所示),并以在所有鉴定结果中均以可信度较高的 Elon-gation Factor 1-alpha 蛋白为例,设置了PTM后,蛋白质序列覆盖率大幅提升。
所有酶解产物的分析在添加PTM之后均有提升,其中 Krakatoa 和 Vesuvius 酶解产物在运行 PEAKS PTM 算法后,序列覆盖率均超过 99%;而胰蛋白酶酶解产物的序列覆盖率仅为 82%。这表明,将半特异性HTA酶与 PEAKS PTM 搜索工具结合使用,可有效提高蛋白质序列覆盖率。
研究结论
PEAKS PTM 算法因无需预先定义PTM类型,是发现非常规胰蛋白酶酶解条件下PTM的理想方法。HTA酶解产物的PTM水平低于胰蛋白酶酶解产物,这一差异大概率源于二者酶解反应条件不同,且HTA酶解过程耗时更短。但在HTA酶解产物中,仍能鉴定出胰蛋白酶酶解产物中不常见的独特PTM(如 pyro-glu form Q)。
针对不同的酶解条件设置最合适的PTM,有助于提高蛋白质序列覆盖率,并最终帮助研究人员获取传统方法可能遗漏的蛋白质信息。
参考文献
1. McCabe M C, Gejji V, Barnebey A, et al. From volcanoes to the bench: Advantages of novel hyperthermoacidic archaeal proteases for proteomics workflows . Journal of Proteomics, 2023, 289: 104992. https://doi.org/10.1016/j.jprot.2023.104992.
2. Xi Han, Lin He, Lei Xin, Baozhen Shan, and Bin Ma Journal of Proteome Research 2011 10 (7), 2930-2936 DOI: 10.1021/pr200153k
3. Dick LW Jr, Kim C, Qiu D, Cheng KC. Determination of the origin of the N-terminal pyro-glutamate variation in monoclonal antibodies using model peptides. Biotechnol Bioeng. 2007 Jun 15;97(3):544-53. doi:10.1002/bit.21260. PMID: 17099914.
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
相关文章
更多 >