

Cell Reports揭示糖尿病肾病新机制:乳酸化修饰TRIM65驱动肾小管损伤
2025-09-22 来源:本站 点击次数:46作为糖尿病主要微血管并发症,糖尿病肾病(DKD)以肾功能渐进性丧失和肾间质纤维化为核心,肾小管上皮细胞(TEC)损伤是疾病进展的关键环节。DKD中TEC存在线粒体功能障碍,导致能量代谢从脂肪酸氧化转向糖酵解,引发乳酸积累;乳酸并非被动代谢产物,而是主动调控TEC损伤的信号分子,但具体机制不明。深圳市人民医院杨书、南昌大学姜玫秀、北京大学深圳医院梁真研究团队在Cell Reports (IF6.9)发文“TRIM65 as a key regulator of ferroptosis and glycolysis in lactate-driven renal tubular injury and diabetic kidney disease”,研究揭示乳酸诱导的TRIM65在K206位点的乳酸化修饰,会损害其在抑制铁死亡和糖酵解中的双重调控作用,进而推动DKD的进展,同时也明确了潜在的治疗靶点。
· 维真助力 ·
病毒产品:AAV9-Ksp1.3-Trim65、AAV9-Ksp1.3-Trim65 K206R、AAV9-Ksp1.3-null
注射方式:肾脏原位注射
· 维真助力 ·
病毒产品:AAV9-Ksp1.3-Trim65、AAV9-Ksp1.3-Trim65 K206R、AAV9-Ksp1.3-null
注射方式:肾脏原位注射
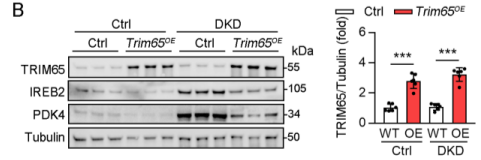
WB检测AAV9-Ksp1.3-Trim65在肾脏中的转染效率
研究结果
1、TRIM65通过泛素化降解IREB2与PDK4抑制铁死亡和糖酵解,对抗糖尿病肾病
单细胞数据库的再分析鉴定出TRIM65主要在肾小管上皮细胞(TEC)中高表达。在糖尿病相关条件刺激下,TEC中IREB2的蛋白及mRNA水平均显著升高,且乳酸对IREB2表达的诱导作用强于HGPA,提示乳酸是调控IREB2的关键代谢信号。内源性及外源性Co-IP等实验证实,TRIM65可与IREB2直接相互作用,并且TRIM65主要通过 K48 型泛素链介导IREB2泛素化。鉴于IREB2是铁死亡的关键调节因子,研究团队探索了TRIM65是否参与铁死亡的调节,发现TRIM65依赖IREB2抑制乳酸诱导的铁死亡,且该调控在乳酸积累的病理状态下更显著。除了与铁死亡相关的基因外,研究团队还发现糖酵解关键调控基因PDK4在TRIM65 KO后蛋白水平显著增加,数据表明PDK4是TRIM65的直接靶标,TRIM65促进PDK4的泛素化和随后的降解。进一步的分析证实TRIM65参与乳酸介导的TEC糖酵解能力上调,且这种调节依赖于PDK4。随后,研究人员探索了TRIM65在DKD进展中的潜在作用,发现Trim65 TKO(肾小管特异性敲除)使糖尿病小鼠的肾损伤恶化,并加重了DKD小鼠肾脏中的脂质积累、氧化应激异常及炎症,此外Trim65 TKO DKD小鼠肾脏中,IREB2和PDK4蛋白水平显著升高,而二者的泛素化水平降低。表明TRIM65缺乏通过促进糖酵解、铁凋亡和炎症加剧DKD,突出了其作为DKD治疗靶点的潜力。
1、TRIM65通过泛素化降解IREB2与PDK4抑制铁死亡和糖酵解,对抗糖尿病肾病
单细胞数据库的再分析鉴定出TRIM65主要在肾小管上皮细胞(TEC)中高表达。在糖尿病相关条件刺激下,TEC中IREB2的蛋白及mRNA水平均显著升高,且乳酸对IREB2表达的诱导作用强于HGPA,提示乳酸是调控IREB2的关键代谢信号。内源性及外源性Co-IP等实验证实,TRIM65可与IREB2直接相互作用,并且TRIM65主要通过 K48 型泛素链介导IREB2泛素化。鉴于IREB2是铁死亡的关键调节因子,研究团队探索了TRIM65是否参与铁死亡的调节,发现TRIM65依赖IREB2抑制乳酸诱导的铁死亡,且该调控在乳酸积累的病理状态下更显著。除了与铁死亡相关的基因外,研究团队还发现糖酵解关键调控基因PDK4在TRIM65 KO后蛋白水平显著增加,数据表明PDK4是TRIM65的直接靶标,TRIM65促进PDK4的泛素化和随后的降解。进一步的分析证实TRIM65参与乳酸介导的TEC糖酵解能力上调,且这种调节依赖于PDK4。随后,研究人员探索了TRIM65在DKD进展中的潜在作用,发现Trim65 TKO(肾小管特异性敲除)使糖尿病小鼠的肾损伤恶化,并加重了DKD小鼠肾脏中的脂质积累、氧化应激异常及炎症,此外Trim65 TKO DKD小鼠肾脏中,IREB2和PDK4蛋白水平显著升高,而二者的泛素化水平降低。表明TRIM65缺乏通过促进糖酵解、铁凋亡和炎症加剧DKD,突出了其作为DKD治疗靶点的潜力。

TRIM65基因敲除加重DKD小鼠肾损伤
2、TRIM65在TECs中的过表达赋予糖尿病小鼠对肾损伤的抵抗力
通过携带肾小管特异性启动子的腺相关病毒,向野生型糖尿病小鼠肾脏TEC中特异性过表达TRIM65,Western blot验证显示AAV9-Trim65转染后,糖尿病小鼠肾脏中TRIM65蛋白水平显著升高,且其下游靶蛋白IREB2和PDK4的蛋白水平降低、泛素化水平升高,证实TRIM65在TEC中成功过表达并恢复了对IREB2和PDK4的泛素化降解功能。不仅如此,TRIM65 过表达显著缓解了糖尿病诱导的肾脏损伤,减少了糖尿病小鼠肾脏的脂质积累和氧化应激,同时降低了糖尿病小鼠肾脏的乳酸含量,下调了糖酵解关键基因的mRNA表达,改善了糖酵解异常;此外,肾脏中炎症相关细胞因子的mRNA水平显著降低,炎症反应得到抑制。机制上,经预测及突变实验证实,研究人员发现TRIM65的K206位点是关键乳酸化位点,TRIM65乳酸化随DKD进展升高,且与IREB2、PDK4蛋白水平正相关、泛素化水平负相关,还与 DKD 严重程度正相关,表明TRIM65 K206对于调节乳酸刺激下的糖酵解反应和铁死亡是关键的。
通过携带肾小管特异性启动子的腺相关病毒,向野生型糖尿病小鼠肾脏TEC中特异性过表达TRIM65,Western blot验证显示AAV9-Trim65转染后,糖尿病小鼠肾脏中TRIM65蛋白水平显著升高,且其下游靶蛋白IREB2和PDK4的蛋白水平降低、泛素化水平升高,证实TRIM65在TEC中成功过表达并恢复了对IREB2和PDK4的泛素化降解功能。不仅如此,TRIM65 过表达显著缓解了糖尿病诱导的肾脏损伤,减少了糖尿病小鼠肾脏的脂质积累和氧化应激,同时降低了糖尿病小鼠肾脏的乳酸含量,下调了糖酵解关键基因的mRNA表达,改善了糖酵解异常;此外,肾脏中炎症相关细胞因子的mRNA水平显著降低,炎症反应得到抑制。机制上,经预测及突变实验证实,研究人员发现TRIM65的K206位点是关键乳酸化位点,TRIM65乳酸化随DKD进展升高,且与IREB2、PDK4蛋白水平正相关、泛素化水平负相关,还与 DKD 严重程度正相关,表明TRIM65 K206对于调节乳酸刺激下的糖酵解反应和铁死亡是关键的。
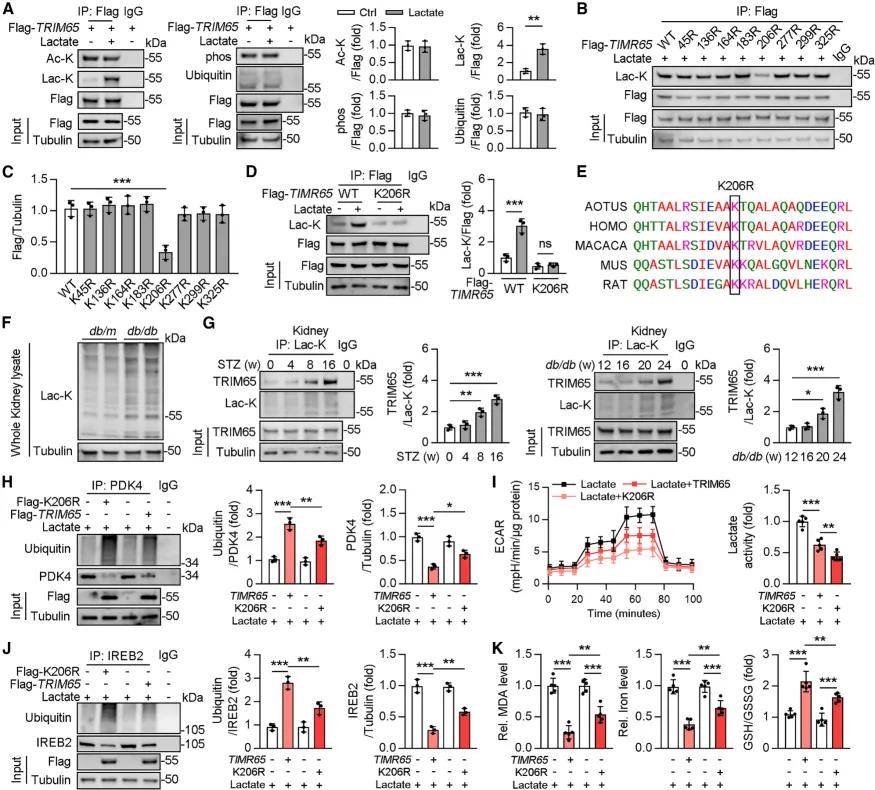
乳酸刺激通过K206乳酸化抑制TRIM65功能
3、乳酸刺激下p300介导TRIM65 K206位点的乳酸化
将AAV9-Trim65和AAV9-Trim65 K206R转染到TRIM65 KO小鼠的肾脏中,并诱导DKD模型。结果显示,野生型TRIM65可逆转TRIM65 KO导致的DKD肾损伤,而TRIM65 K206R突变体对DKD的保护效果更显著;同时,两种TRIM65均可下调KO小鼠肾脏中IREB2和PDK4的蛋白水平,证实 TRIM65的K206乳酸化是小鼠DKD发病的关键因素,该位点乳酸化缺失可增强TRIM65的抗DKD作用。进一步的探索分析发现,p300(乳酸转移酶)可与TRIM65直接结合(结合区域为TRIM65的A139-260位氨基酸)。在乳酸刺激下,糖尿病模型小鼠肾脏中p300蛋白水平升高,且乳酸会增强二者相互作用;p300过表达能提高TRIM65乳酸化水平,p300抑制剂则可抑制该乳酸化,且 TRIM65 K206R突变会阻断p300对TRIM65乳酸化的促进作用,证实p300是介导乳酸诱导TRIM65 K206位点乳酸化的关键分子。
将AAV9-Trim65和AAV9-Trim65 K206R转染到TRIM65 KO小鼠的肾脏中,并诱导DKD模型。结果显示,野生型TRIM65可逆转TRIM65 KO导致的DKD肾损伤,而TRIM65 K206R突变体对DKD的保护效果更显著;同时,两种TRIM65均可下调KO小鼠肾脏中IREB2和PDK4的蛋白水平,证实 TRIM65的K206乳酸化是小鼠DKD发病的关键因素,该位点乳酸化缺失可增强TRIM65的抗DKD作用。进一步的探索分析发现,p300(乳酸转移酶)可与TRIM65直接结合(结合区域为TRIM65的A139-260位氨基酸)。在乳酸刺激下,糖尿病模型小鼠肾脏中p300蛋白水平升高,且乳酸会增强二者相互作用;p300过表达能提高TRIM65乳酸化水平,p300抑制剂则可抑制该乳酸化,且 TRIM65 K206R突变会阻断p300对TRIM65乳酸化的促进作用,证实p300是介导乳酸诱导TRIM65 K206位点乳酸化的关键分子。
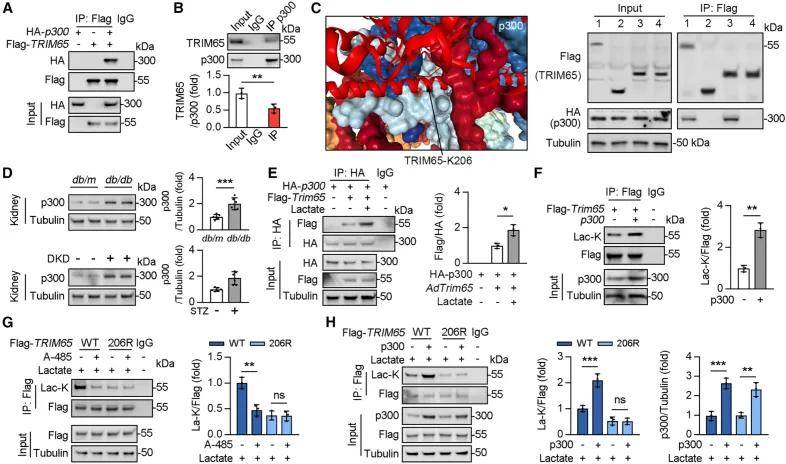
乳酸刺激下p300介导TRIM65 K206位点的乳酸化
研究结论
本研究表明乳酸通过催化TRIM65蛋白K206位点的乳酸化修饰,抑制其泛素连接酶活性。这一修饰阻止了铁调节蛋白IREB2和糖酵解关键基因PDK4的降解,从而在DKD中驱动铁死亡和糖酵解过程。研究同时发现,无法被乳酸化的K206R突变体能显著增强抗DKD疗效,由此确立TRIM65乳酸化作为治疗DKD的新靶点。
本研究表明乳酸通过催化TRIM65蛋白K206位点的乳酸化修饰,抑制其泛素连接酶活性。这一修饰阻止了铁调节蛋白IREB2和糖酵解关键基因PDK4的降解,从而在DKD中驱动铁死亡和糖酵解过程。研究同时发现,无法被乳酸化的K206R突变体能显著增强抗DKD疗效,由此确立TRIM65乳酸化作为治疗DKD的新靶点。
相关文章
更多 >