

AI在药物研发中的演进及其在虚拟筛选与化合物合成中的应用和案例
2025-11-20 来源:本站 点击次数:613在药物发现中,传统湿实验具有实验周期长、测试成本高、苗头物命中率低的特点[1]。计算工具凭借算法的快速进步,在降低药物发现的成本与风险的同时显著提升了研究通量,已成为药物开发的关键手段[2]。人工智能 (Artificial Intelligence,AI) 在分析复杂生物系统、识别疾病生物标志物与潜在药物靶点、模拟药物-靶点相互作用、预测化合物安全性/有效性和优化临床试验管理等方面显示出重要价值[3]。
Section.01
药物研发中的 AI
近年来,基于 AI 的生物分析算法发展迅猛。这些算法通过构建模拟人类智能的系统,可高效处理生物网络数据,实现分类、聚类及预测等功能。凭借该能力,AI 可通过基因互作网络解析癌症的复杂性,深化对致癌机制的理解并揭示新型抗癌靶点[4]。自 2018 年起,AI 在制药领域的应用已从概念阶段 ("0") 迈入实际应用阶段 ("1")。2024 年,诺贝尔物理学奖授予了机器学习 (ML) 中人工神经网络的开创性进展——该技术正是驱动当前深度学习 (DL)、自然语言处理 (NLP) 和计算机视觉等 AI 方法的核心基础[5]。
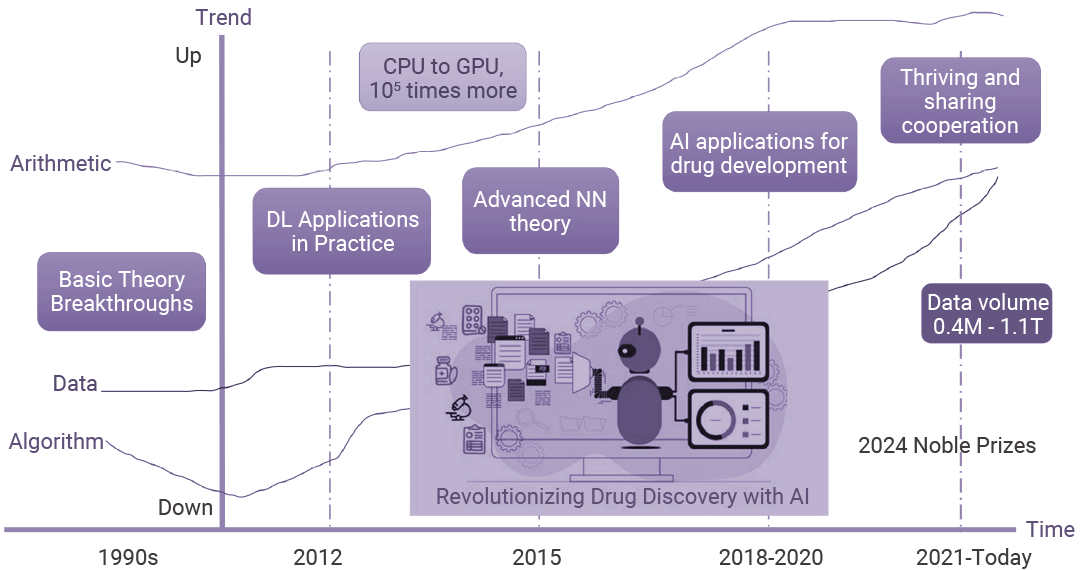 图 1. AI 在药物研发中的技术发展简述[5]。
图 1. AI 在药物研发中的技术发展简述[5]。如今,AI 分析海量数据的能力正在重塑药物开发格局。从靶点识别、药物发现、临床前研究、临床试验到监管审评与上市后监测,AI 技术在整个研发链条中均展现出显著优势。这一变革潜力已促使制药企业、生物技术公司和研究机构广泛采用 AI 技术,以突破传统方法的局限[3]。
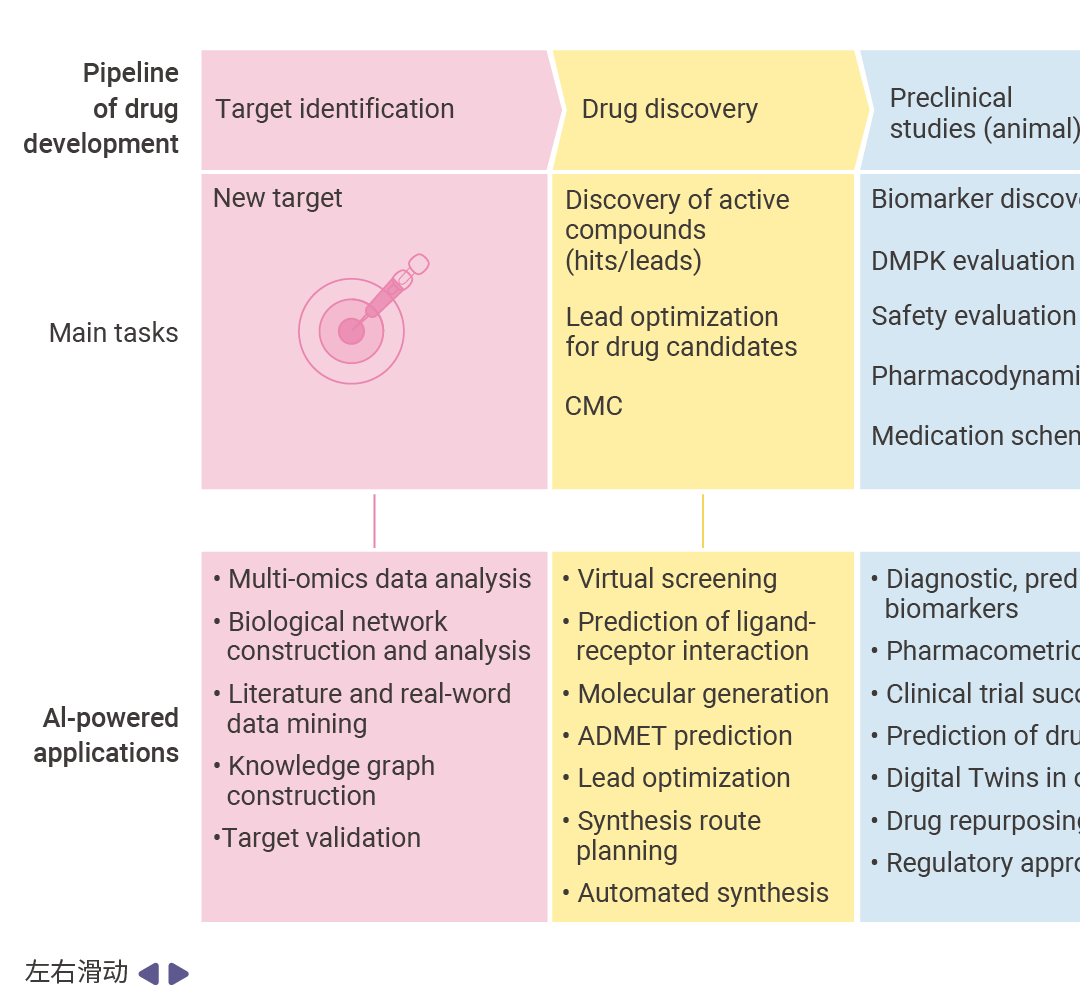
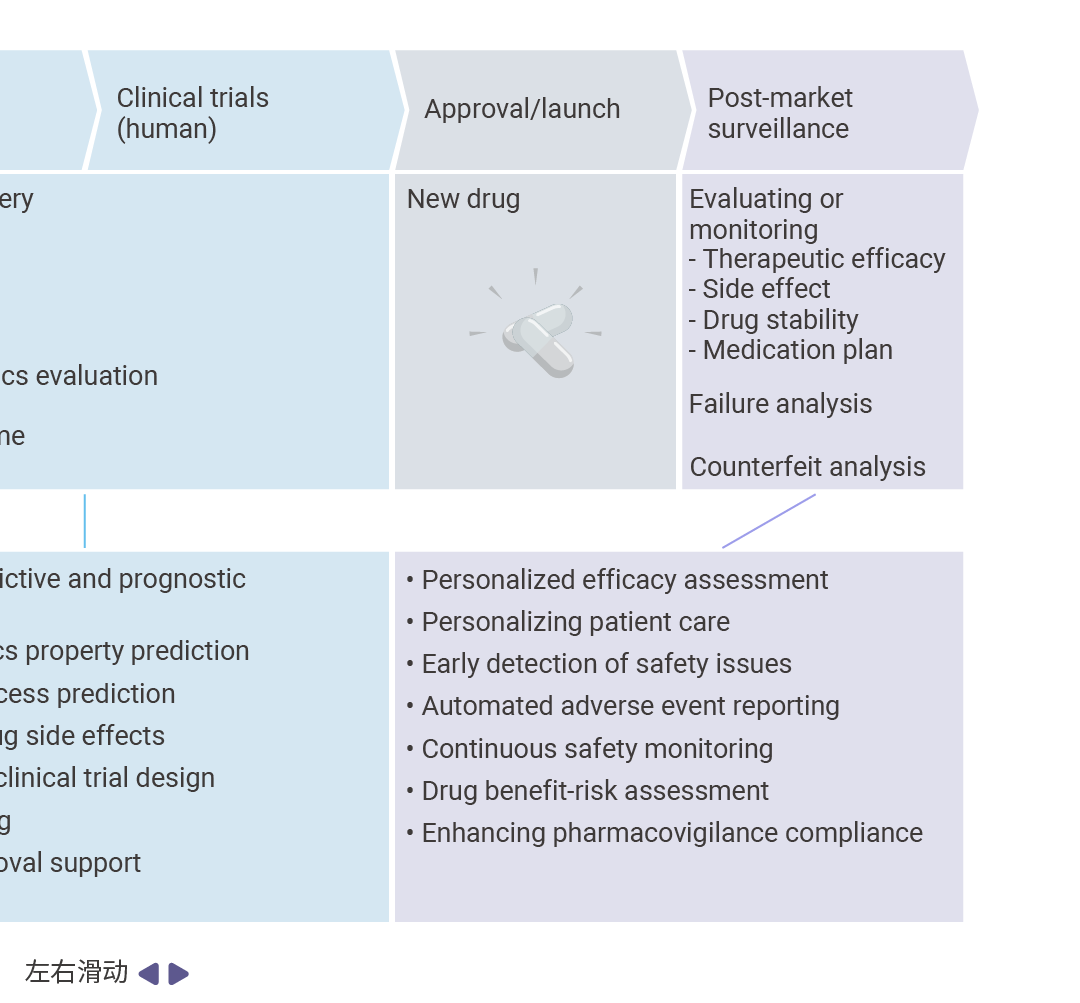
图 2. AI 在药物研发管线中的应用概述[3]。
Section.02
AI 与药物研发:多元化应用
AI 在虚拟筛选中的应用
虚拟筛选通过计算分析大型化合物库,以识别对特定生物靶点具有高结合潜力的化合物。机器学习 (ML) 模型为基于配体的虚拟筛选 (LBVS) 提供了强大助力,其中定量构效关系 (QSAR) 模型利用已知的配体特性来预测新的候选化合物。近年来,随着新型分子表征方法和深度学习 (DL) 架构的发展,QSAR 领域迎来了 AI 革命。深度 QSAR 技术现已能够高效筛选超大规模化合物库,并常与药效团建模或分子对接技术结合使用。
分子对接技术则利用蛋白质三维结构来识别潜在抑制剂,构成了基于蛋白结构的虚拟筛选 (SBVS) 的基础。AI 的进步优化了分类方法、结合口袋发现以及用于评估配体-蛋白质亲和力的评分函数。新兴的基于 DL 的评分函数,特别是卷积神经网络 (CNN) 模型正在虚拟筛选中崭露头角,其通过处理海量数据并识别与靶标有效结合相关的结构特征而获得广泛应用[6]。
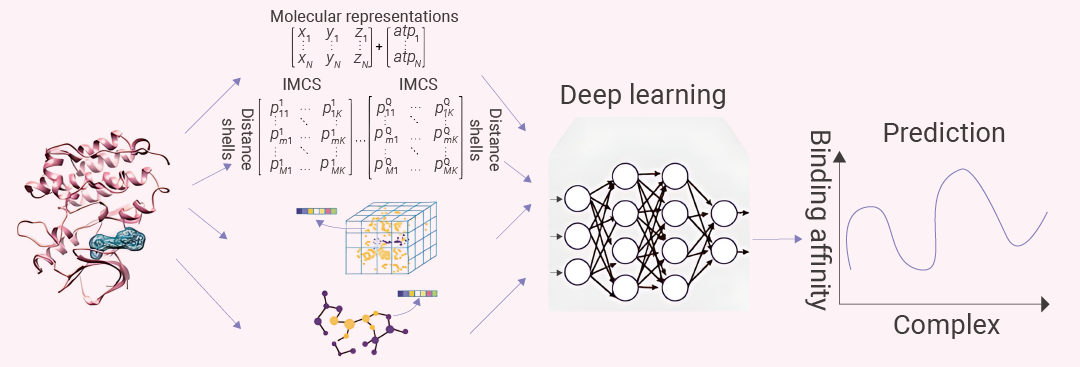 图 3. 基于 DL 模型预测蛋白和小分子结合的流程图[7]。
图 3. 基于 DL 模型预测蛋白和小分子结合的流程图[7]。AI 驱动的化合物合成规划
在小分子药物研发中,化学合成仍是主要瓶颈之一,这一过程技术含量高且极为耗时费力。计算机辅助合成规划 (CASP) 和有机化合物的自动化合成能够帮助化学家从重复性劳动中解放出来,使其能够专注于更具创新性的工作。
现代 CASP 工具基于早期采用逻辑启发式的规则系统,通过逆合成分析有效确定最佳反应路径。最新突破显示,Transformer 模型已成功应用于合成规划的关键环节,包括逆合成分析、区域选择性和立体选择性预测,以及反应指纹提取[3]。
尽管纯数据驱动的 AI 方法最初在复杂合成规划的可靠性方面引发担忧,但这一挑战推动了稳健混合系统的开发——将 AI 与成熟的化学规则智能结合。RetroExplainer 它提出了一种可解释的深度学习框架,将逆合成概念化为分子组装过程。这一创新方法不仅展现出优于传统方法的性能,还通过定量归因分析实现了前所未有的可解释性,使决策过程透明化[8]。
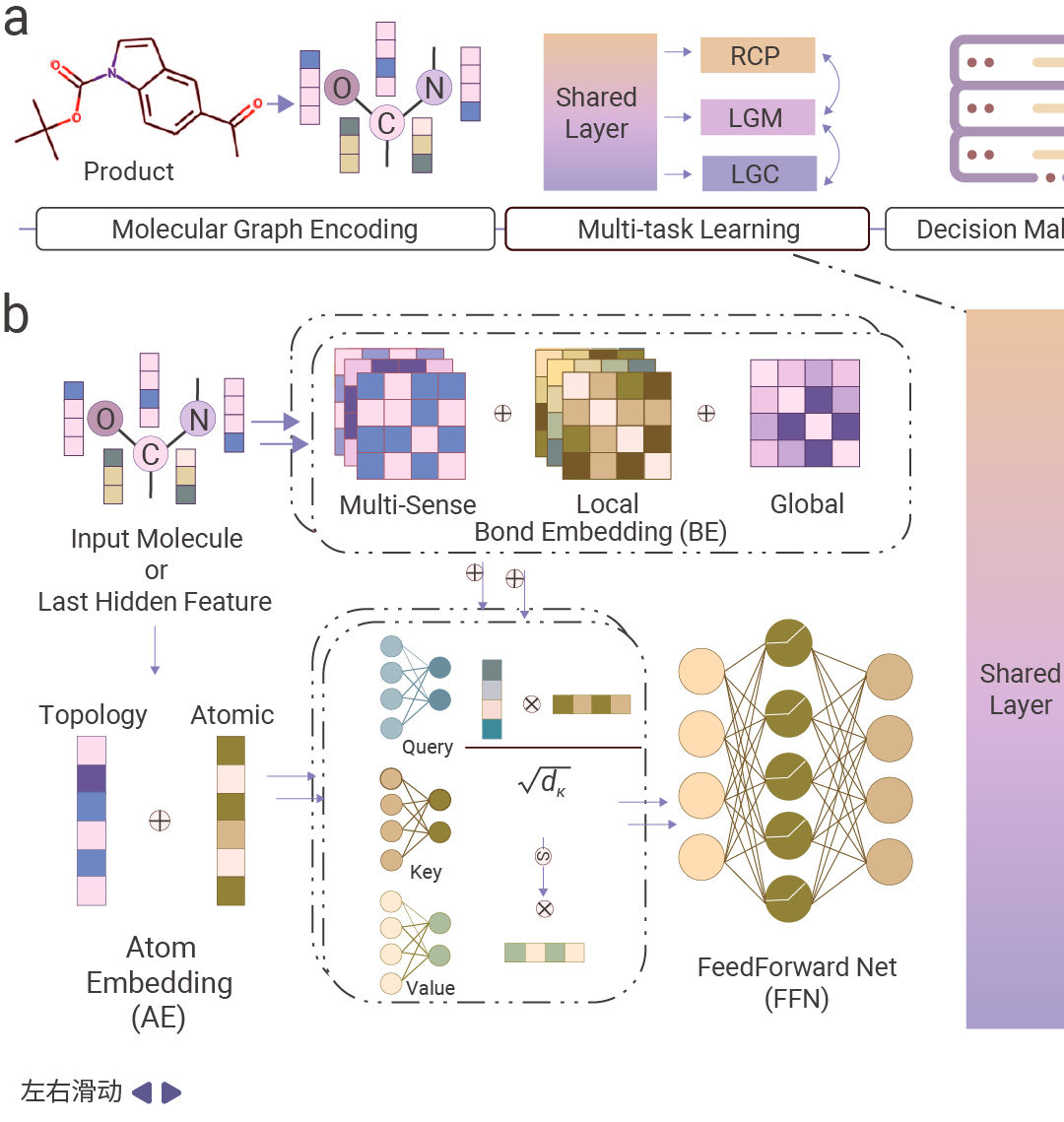
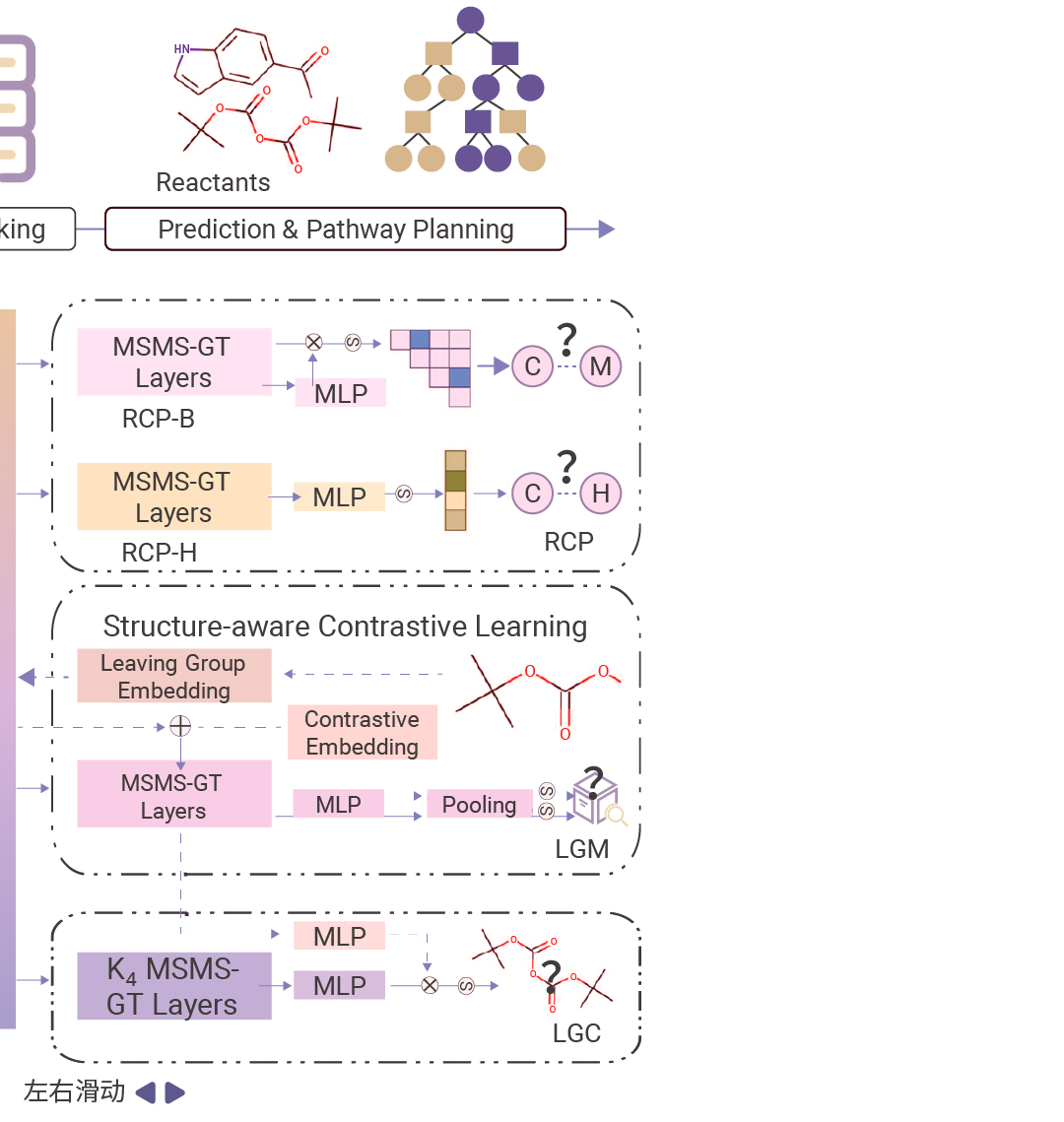
图 4. RetroExplainer 流程概述[8]。
Section.03
AI 药物发现:研究案例
GeminiMol DL 模型加速大规模药物发现
GeminiMol 将构象空间特征融入分子表征学习,旨在捕捉分子结构与构象空间之间的复杂关联。该模型在 67 个分子特性预测、73 个细胞活动预测和 171 个零样本任务 (包括虚拟筛选和靶标识别) 中,利用构象信息的模型显示出优于传统表征方法的性能[9]。基于分子构象空间相似性描述符的对比学习框架是训练分子表征模型的有效策略,该策略能够快速探索化学空间并促进新的药物发现范式。
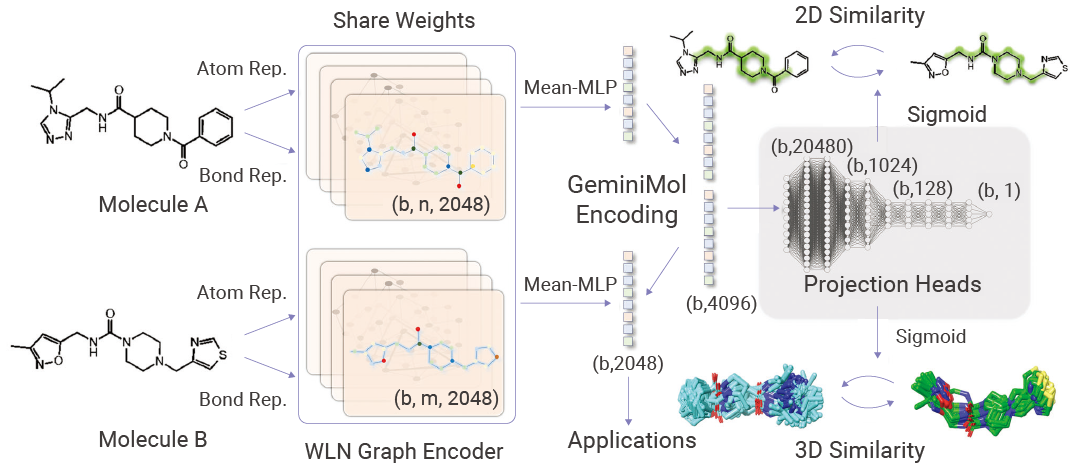 图 5. 通过分子间对比学习框架训练分子构象空间表征模型 GeminiMol[9]。
图 5. 通过分子间对比学习框架训练分子构象空间表征模型 GeminiMol[9]。虚拟筛选驱动的 MYH9 抑制剂的高效鉴定
研究人员通过计算机虚拟筛选技术初步鉴定出可与 Human MYH9 蛋白特异性结合的小分子化合物。随后采用 CCK-8 细胞增殖 (HY-K0301) 检测法系统评估了这些化合物对原代小鼠软骨细胞增殖活性的影响,并运用 β- 半乳糖苷酶染色法 (HY-K1089) 检测其对细胞衰老进程的调控作用。在此基础上,研究团队进一步建立了小鼠骨关节炎 (OA) 模型,对这些化合物的治疗效果进行了体内验证。最终研究发现,4,5-dicaffeoylquinic acid 能够有效阻断 DPP4 与 MYH9 之间的蛋白相互作用,显著改善由 DPP4 过表达所导致的小鼠创伤后骨关节炎及衰老相关性骨关节炎的病理进程[10]。
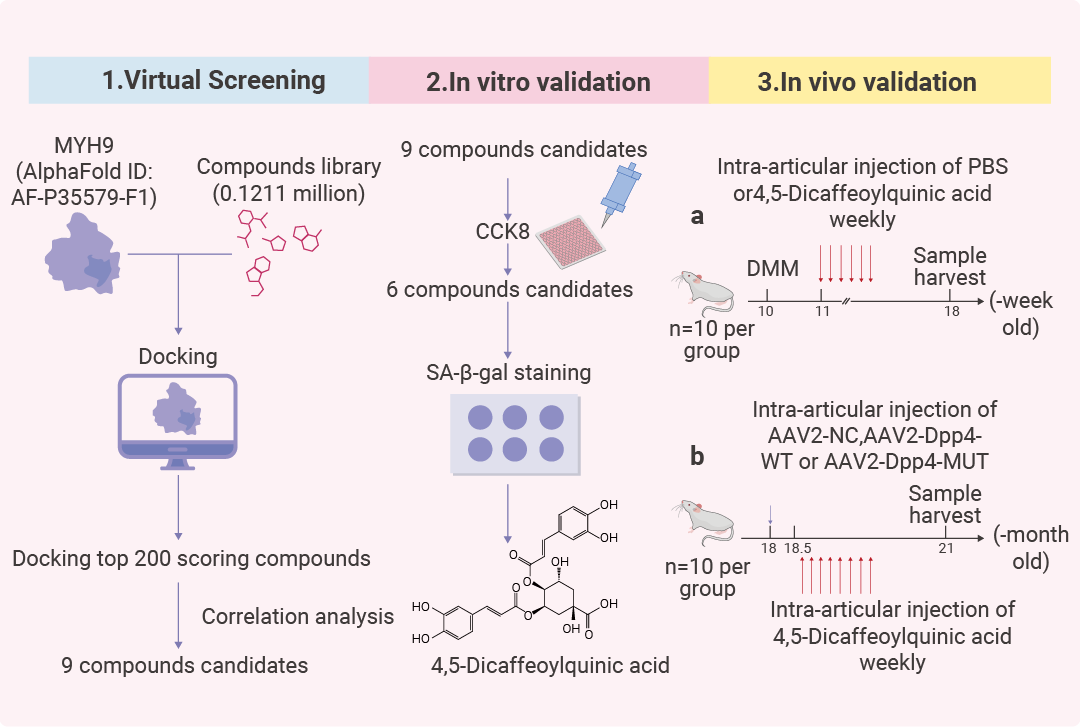 图 6. 靶向人 MYH9 的药物筛选方案[10]
图 6. 靶向人 MYH9 的药物筛选方案[10]MCE AI 驱动药物筛选平台
虚拟筛选通常依赖于计算机模拟和分子对接技术,通过计算分子间的相互作用来预测化合物的生物活性。AI 药物筛选是一种结合 AI 技术与计算化学的高通量筛选方法,广泛应用于蛋白结构预测、新药研发和分子设计与优化等领域。其主要目的是利用机器学习 (Machine Learning,ML) 算法分析大量数据,从中学习规律,生成 AI 打分函数,以此提高筛选效率,加速候选药物的发现过程。
MCE AI 药物筛选平台综合使用分子对接、机器学习、分子动力学模拟等方法,借助高性能服务器,能够在最短数小时内完成数千万分子的筛选,真正实现快速、高效!
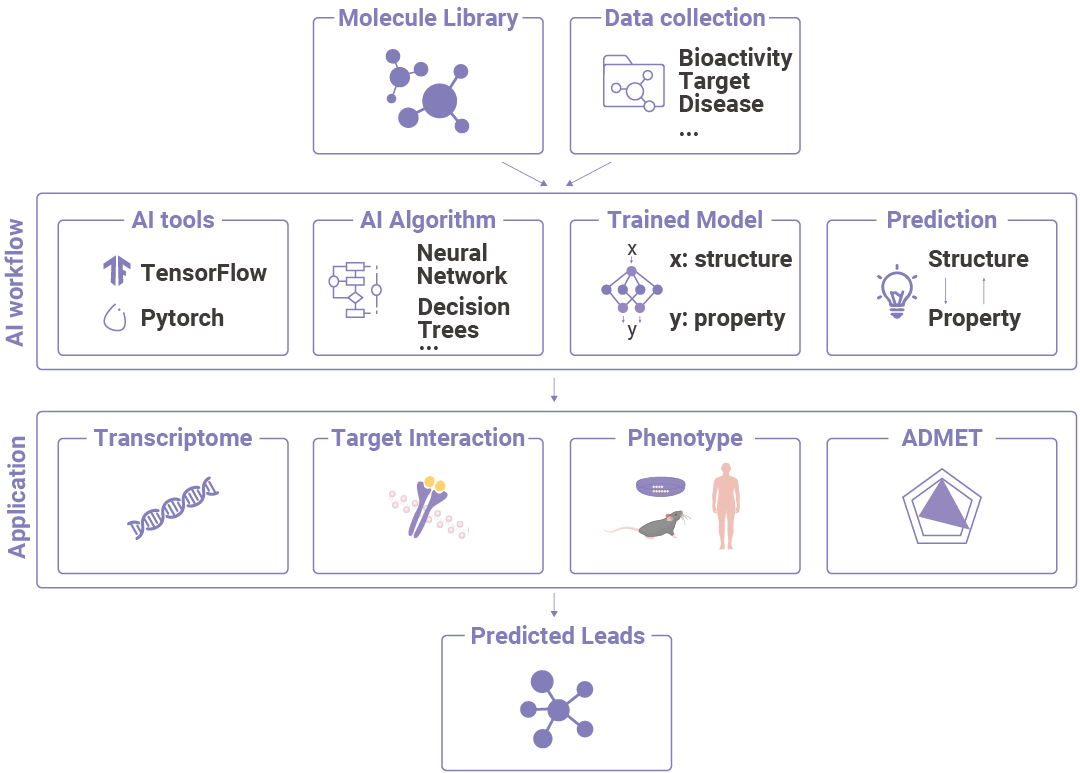 图 7. AI 技术在药物发现中的应用。
图 7. AI 技术在药物发现中的应用。Section.04
小结
总体而言,AI 是强大的“加速器”,AI 技术的不断进步正在大大提高药物开发的效率。但生物学系统的复杂性决定了实验验证仍是金标准。未来的突破将依赖于“干湿结合”(AI+ 自动化实验) 的协同迭代,而非单一技术的替代。
|
产品推荐 |
|
DNA 编码化合物库 (DNA Encoded compound Library,DEL) 技术作为新颖、强大的苗头化合物发现引擎,可快速从几千万至数十亿分子中,遴选出结构新颖、具有潜在成药性的化合物,大大缩短药物研究周期,降低研发成本。在 DEL 库中,每一个分子砌块 (Building Block) 都由一段已知唯一的 DNA 序列进行标记,通过 DNA 兼容反应和组合化学模式,历经数个循环即可获得上亿 DEL 分子。数十亿化合物可以混合在一管中筛选,最终通过高通量测序技术,解码 DEL 分子的专属 DNA 标签,快速获得针对靶点的苗头化合物信息。 |
|
50K Diversity Library (HY-L901) 由 50,000 种类药化合物组成。本多样性库具备新颖性、类药性,结构多样性等特点,库中化合物可重复供应,是新药研发的有力工具,可以广泛地应用于高通量筛选 (HTS) 和高内涵筛选 (HCS)。 |
|
由 5,000 种类药化合物组成,每种化合物代表一种结构骨架,最大程度保证了库的结构多样性。库中的化合物均经过 MedChem & PAINS filters 筛选,剔除了不合适的化学结构,避免“目标错误”。本库化合物数量少但结构足够多样,是药物筛选的有力工具。 |
|
3D Diverse Fragment Library (HY-L903) 由 5,196 个非平面片段分子组成 (平均 Fsp3 值为 0.58),超过 4,700 个片段至少包含一个手性中心。本库设计的关键元素是 3D 结构、多样性、生物反应性等,有效提高了片段潜在生物活性,为基于片段的药物发现提供了更高的片段命中概率。 |
|
Drug Fragment Library (HY-L904) MCE Drug Fragment Library 由 1,000 个药物片段组成。这些药物片段来自 2,946 个 FDA 已批准的药物分子,同一药物的不同片段可以出现在其他药物中,这些片段和 PK/PD 性质存在一定的相关性,基于片段的筛选可以为后续优化结构预留出足够的化学空间,该化合物库是 FBDD(基于片段的药物设计)药物筛选的必备工具。 |
|
Natural Product-like Library (HY-L905) MCE Natural Product-like Compound Library 由 5,000 个来自类药库的类天然产物化合物组成,库中每个分子含有天然产物关键骨架 (42 个) 或者和天然产物的谷本相似系数大于 0.6,且Natural-likeness scoring > -2,该化合物库同时具备类药性和新颖性,库中化合物可重复供应,是新药研发的有力工具,可以广泛地应用于高通量筛选 (HTS) 和高内涵筛选 (HCS)。 |

[1] Chen W, et al. Artificial intelligence for drug discovery: Resources, methods, and applications. Mol Ther Nucleic Acids. 2023 Feb 18;31:691-702.
[2] Wu Y, et al. The role of artificial intelligence in drug screening, drug design, and clinical trials. Front Pharmacol. 2024 Nov 29;15:1459954.
[3] Zhang K, et al. Artificial intelligence in drug development. Nat Med. 2025 Jan;31(1):45-59.
[4] You Y,et al. Artificial intelligence in cancer target identification and drug discovery. Signal Transduct Target Ther. 2022 May 10;7(1):156.
[5] Chen Fu et al. Journal of Pharmaceutical Analysis, 2025, 101248, ISSN 2095-1779.
[6] Ocana A, et al. Integrating artificial intelligence in drug discovery and early drug development: a transformative approach. Biomark Res. 2025 Mar 14;13(1):45.
[7] Wu J, et al. Large-scale comparison of machine learning methods for profiling prediction of kinase inhibitors. J Cheminform. 2024 Jan 30;16(1):13.
[8] Wang Y, et al. Retrosynthesis prediction with an interpretable deep-learning framework based on molecular assembly tasks. Nat Commun. 2023 Oct 3;14(1):6155.
[9] Wang L, et al. Conformational Space Profiling Enhances Generic Molecular Representation for AI-Powered Ligand-Based Drug Discovery. Adv Sci (Weinh). 2024 Oct;11(40):e2403998.
[10] Li X, et al. Dipeptidyl Peptidase 4 (DPP4) Exacerbates Osteoarthritis Progression in an Enzyme-Independent Manner. Adv Sci (Weinh). 2025 Feb;12(6):e2410525.

