

文献解析:免疫肽组学解锁微环境驱动的 1 型糖尿病核心新抗原
2026-04-02 来源:本站 点击次数:5051 型糖尿病是一种由自身免疫攻击胰岛 β 细胞引发的慢性疾病,尽管学界对胰岛素等天然抗原的研究已十分深入,但新型自身抗原的产生机制始终是未解之谜。近年来,肿瘤免疫学研究发现,慢性炎症可通过非突变机制产生 “新抗原”,这一思路为自身免疫病研究提供了全新方向。本研究通过免疫肽组学技术,在人类和小鼠体内发现了一种由微环境驱动的胰岛素单氨基酸突变——C19S(半胱氨酸→丝氨酸),它不仅会重塑胰岛素的自身反应性,还会成为持续激活记忆 T 细胞的关键 “新抗原”,为 1 型糖尿病的发病机制和诊疗研究打开了新窗口。
发表时间:2026年1月
期刊:Nature Immunology (IF:27.7)
文章标题:A microenvironment-driven HLA-II-associated insulin neoantigen elicits persistent memory T cell activation in diabetes
核心内容:本研究依托免疫肽组学精准技术,首次在人类和小鼠体内系统鉴定了 1 型糖尿病相关的胰岛免疫肽谱。研究突破性地发现,在应激微环境诱导下,胰岛素B 链第19位氨基酸发生关键的C19S(半胱氨酸→丝氨酸)单氨基酸突变,并形成大量可被 MHC-II 分子提呈的新型免疫原性肽段。这类 “C19S 新抗原” 不仅能打破免疫耐受,还能驱动 CD4+ T 细胞克隆扩增与持续活化,从根本上解释了1型糖尿病中慢性炎症的维持机制。该发现通过免疫肽检测视角,重新定义了 1 型糖尿病的致病抗原谱,为开发精准诊断标志物与免疫治疗新靶点提供了核心科学依据。

研究方法
本研究基于免疫肽组学技术体系,首先对健康对照(ND)、发病 3 个月(3mos)及 18 个月(18mos)的 1 型糖尿病患者进行混合餐耐量试验(MMTT),采集不同时间点外周血单核细胞,通过优化 HLA-II 免疫沉淀、LC-MS/MS等实验流程,结合数据非依赖性质谱(DDA)与靶向质谱(PRM),在人类受试者及 NOD 小鼠模型的外周血/淋巴组织中精准鉴定并验证了应激微环境下的关键免疫肽段 ——C19S 突变胰岛素肽段;随后利用 ELISpot、T 细胞杂交瘤/四聚体染色、光谱流式及单细胞 RNA 测序等技术,从跨物种保守性、免疫原性、疾病相关微环境依赖性及T细胞功能特征等维度,系统验证了该“新抗原”的形成机制、免疫原性及与1型糖尿病病程的相关性,并结合临床患者样本分析了其转化潜力。
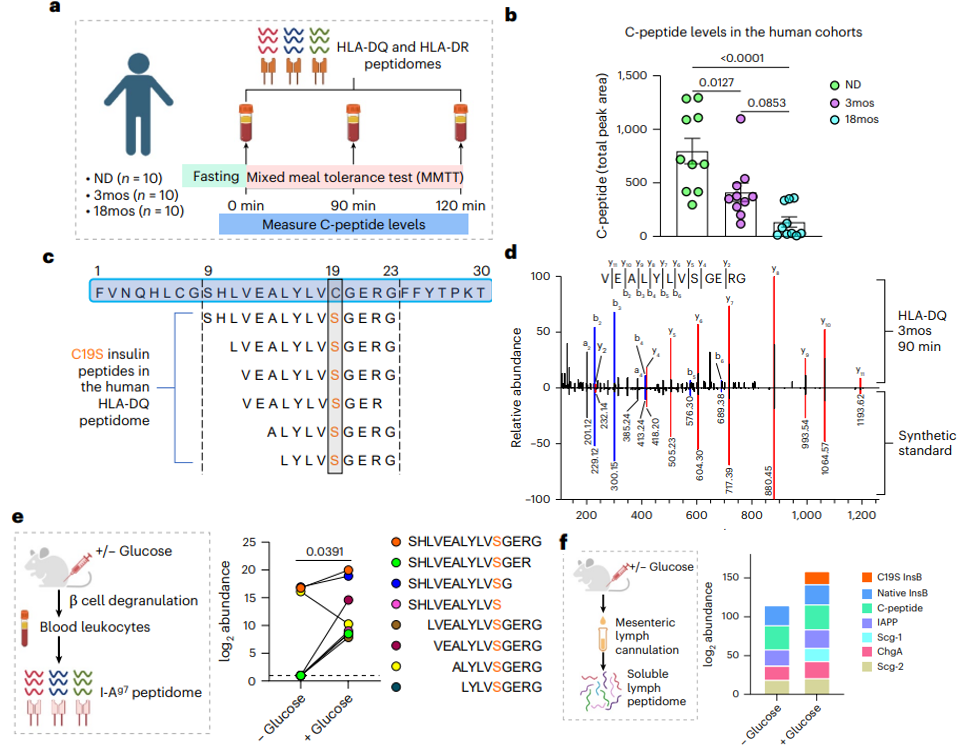
图1免疫肽组学鉴定并验证1型糖尿病相关C19S新抗原
研究结果1. 小鼠和人类中发生半胱氨酸 - 丝氨酸突变的胰岛素肽段的鉴定
采用免疫肽组学分析,对人类受试者(健康对照、1 型糖尿病发病3个月/18 个月患者)进行混合餐耐量试验(MMTT),同时对NOD小鼠进行葡萄糖刺激,通过质谱技术筛选并验证胰岛来源肽段。在人类 HLA-DQ/DR 肽组和小鼠 I-Ag7 肽组中,均鉴定出胰岛素 B 链第 19 位半胱氨酸突变为丝氨酸的 C19S 肽段,该肽段覆盖 InsB9-23区域(MHC-II 结合核心区),仅在葡萄糖/MMTT刺激后可稳定检测;进一步通过合成标准品质谱比对,确证了C19S肽段在人类外周血、小鼠淋巴液及胰岛组织中的真实存在,证实其跨物种保守性。
2. 疾病相关信号促进胰腺 β 细胞中 C19S 的产生
构建 C19S 特异性 CD4⁺T 细胞杂交瘤(S5 克隆),建立基于 β 细胞颗粒的抗原提呈(GAP),结合内质网应激剂(衣霉素)、炎症细胞因子(TNFα、IL-1β 等)处理 MIN6 细胞、小鼠原代胰岛及人类胰岛,检测杂交瘤细胞的激活程度,同时用质谱验证,量化 C19S 肽段的产生水平。内质网应激和氧化应激可显著诱导 β 细胞中 C19S 肽段生成,且该效应可被抗氧化剂(谷胱甘肽、NAC)逆转;炎症细胞因子(尤其是 TNFα)通过加重内质网应激和 redox 应激,进一步增强 C19S 产生;随着糖尿病进展,NOD 小鼠胰岛细胞对 C19S 特异性 T 细胞的活化效应逐渐升高,证实 C19S 提呈水平与疾病进程正相关。
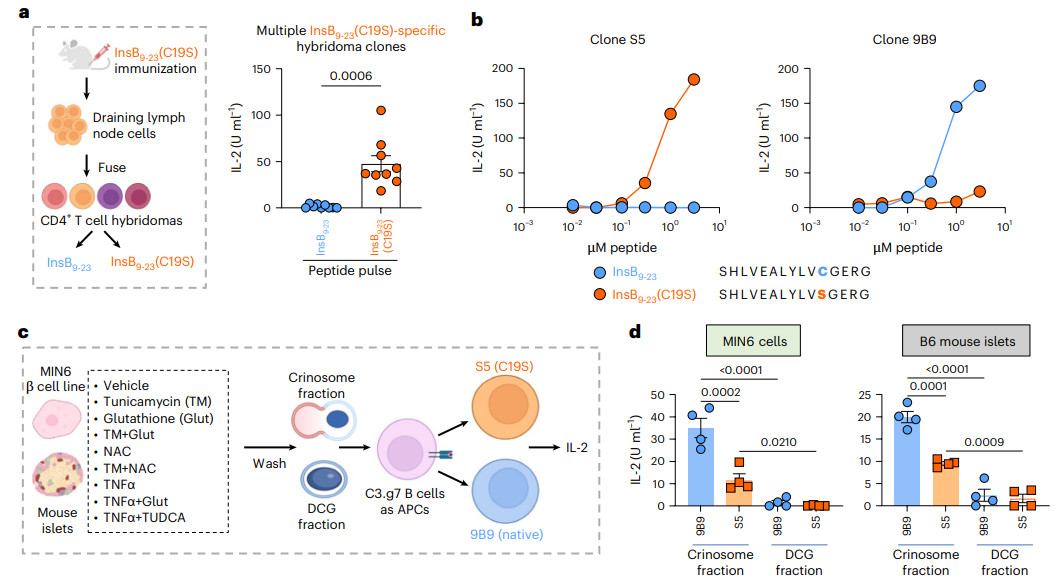
图 2 C19S 特异性 CD4⁺T 细胞杂交瘤构建及应激诱导 C19S 产生的验证
3. C19S 是一种依赖于环境的单氨基酸突变对衣霉素处理前后的 MIN6 细胞分泌溶酶体肽组进行无偏倚质谱分析,通过 Peaks Studio检索,系统筛选翻译后修饰(PTMs)和单氨基酸变异体(SAVs),量化不同条件下 C19S 的丰度变化。在 348 种 PTMs/SAVs 中,C19S(Cys→Ser)在衣霉素处理后增幅最显著,且被鉴定为 SAV 而非传统 PTMs;非应激状态下,C19S 在 β 细胞中仅以低基础水平存在于分泌溶酶体中,而应激微环境可使其成为胰岛素 B 链第 19 位残基的主导修饰形式,证实其环境依赖性特征。
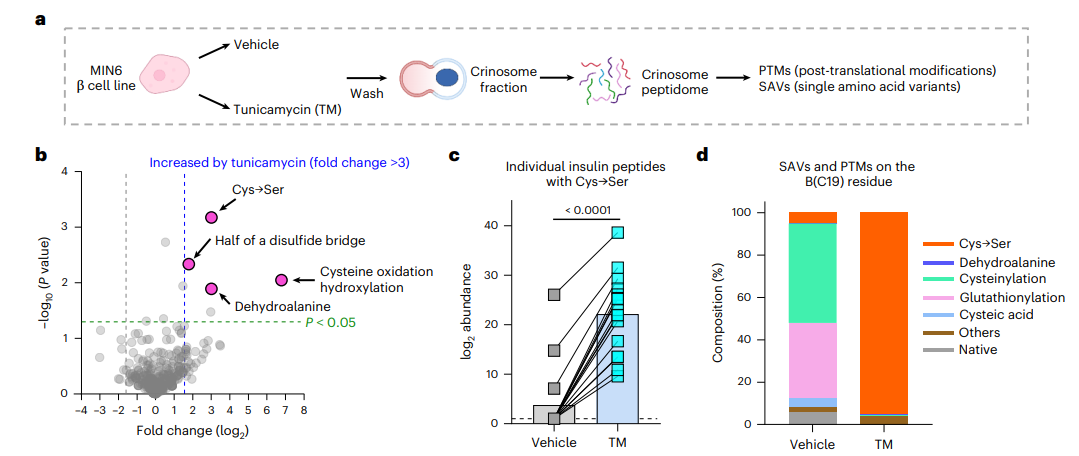
图3. 内质网应激诱导胰岛 β 细胞产生 C19S 单氨基酸突变
4. C19S 新表位的可及性和炎症信号驱动特异性 T 细胞活化
利用 NOD.B16A(胰岛素 Y16A 突变)和 NOD.Tnfrsf1a/1b⁻/⁻(TNF 受体双敲除)小鼠模型,结合 ELISPOT、流式细胞术,分析 C19S 表位可及性和 TNFα 信号对 T 细胞活化的影响。NOD.B16A 小鼠中 C19S 表位缺失,导致 C19S 特异性 T 细胞频率和活化水平显著降低,证实表位可及性是 T 细胞活化的前提;TNFα 信号缺陷可抑制 C19S 特异性 T 细胞的活化表型;即使 β 细胞丢失导致抗原产生减少,糖尿病炎症微环境仍能维持 C19S 特异性 T 细胞的高活化状态。
5. 应激和炎症状态下人类胰岛中的 C19S 突变对非糖尿病供体人类胰岛进行衣霉素(内质网应激)、炎症细胞因子(TNFα、IL-1β、IFNγ)处理,通过 GAP assay 和质谱分析,检测 C19S 肽段的产生情况。衣霉素处理可显著增加人类胰岛分泌溶酶体和致密核心颗粒中 C19S 的水平,抗氧化剂可逆转该效应;IL-1β、TNFα、IFNγ 等炎症细胞因子联合刺激后,人类胰岛分泌溶酶体肽组中可检测到 C19S 胰岛素肽段,证实应激和炎症信号同样可驱动人类胰岛产生 C19S 突变。
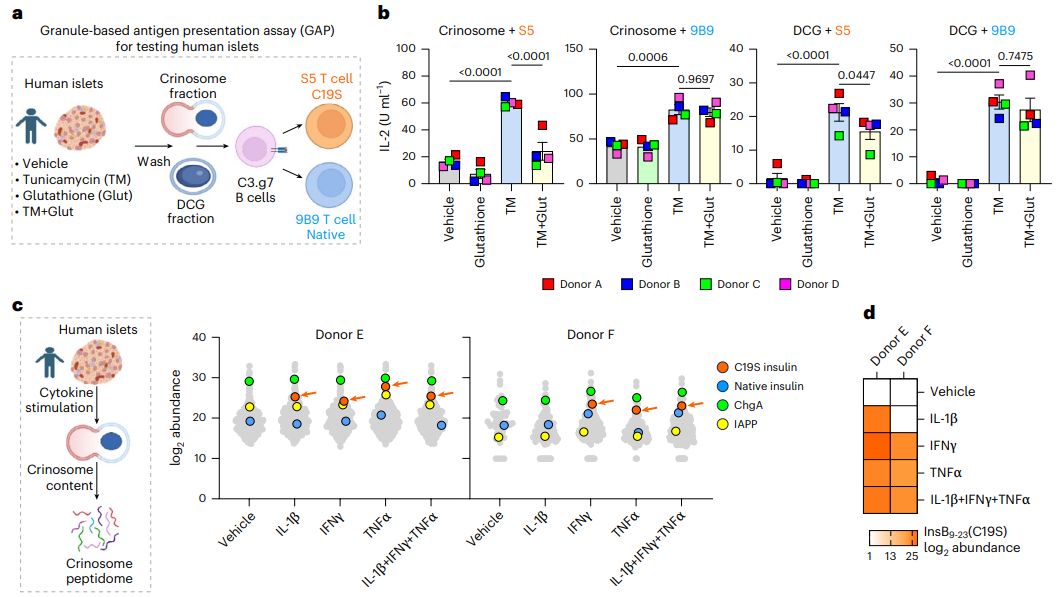
图 4 人类胰岛中应激与炎症信号驱动 C19S 突变的验证
结论本研究通过免疫肽组学技术,在人类与小鼠模型中首次鉴定出微环境驱动的胰岛素 C19S 单氨基酸突变新抗原:该突变由胰岛 β 细胞氧化应激、内质网应激及炎症因子(如 TNFα)诱导产生,在应激状态下成为胰岛素 B 链第 19 位残基的主导修饰形式;C19S 新抗原以登记依赖方式重塑 CD4⁺T 细胞识别,在 1 型糖尿病患者中可诱导 HLA-DQ8 限制性特异性 T 细胞显著扩增,并表现出持续性 Th1 样中枢记忆活化表型,缺乏调节功能且与疾病进展高度相关,为解析 1 型糖尿病自身免疫慢性炎症的维持机制、开发精准诊断标志物与免疫治疗靶点提供了核心科学依据。
参考文献Srivastava, N., Vomund, A.N., Yu, R. et al. A microenvironment-driven HLA-II-associated insulin neoantigen elicits persistent memory T cell activation in diabetes. Nat Immunol 27, 82–97 (2026).