

RWPE-1人正常前列腺上皮细胞专用培养基助力揭示新的药物耐药机制
2026-04-09 来源:本站 点击次数:488
中乔新舟助力相关成果发表
 我们荣幸地向长期使用中乔新舟无血清培养基的合作伙伴致以热烈祝贺
我们荣幸地向长期使用中乔新舟无血清培养基的合作伙伴致以热烈祝贺
由重庆医科大学研究团队完成的成果《KDM5B-driven glucose metabolic reprogramming promotes enzalutamide resistance in prostate cancer via the lactate/hnRNPA1 lactylation/AR-V7 axis》发表于国际知名期刊《Molecular Cancer》(IF=33.9)。在该研究中,作者团队选用了中乔新舟提供的RWPE-1人正常前列腺上皮细胞专用培养基(货号:ZM0351),用于关键细胞模型的培养。
该研究确定了一种机制,将代谢、表观遗传学和KDM5B/AR反馈环与耐药性联系起来。这些发现表明,多靶点策略可能是克服CRPC中Enza耐药性的有希望的方法。
一、研究背景与科学问题
前列腺癌(PCa)仍然是全世界男性癌症相关死亡的主要原因之一。其发病率在不同地区有显著差异。雄激素受体信号抑制剂(ARSIs),如恩扎鲁胺(Enza),是晚期PCa的主要治疗方法。这些药物通过阻断雄激素受体(AR)信号来抑制肿瘤生长。然而,大多数患者最终发展为去势抵抗型前列腺癌(CRPC)对ARSIs的抗性涉及复杂的机制,其中一个关键因素是组成性活性雄激素受体剪接变异体(AR-Vs)的发展。最著名的变体AR-V7缺乏Enza所针对的配体结合域(LBD)。这种缺失使 AR-V7 持续活跃,即使在强效 ARSI 存在的情况下,也能促进亲肿瘤的转录程序。虽然AR-V7的下游效应已被充分描述,但控制其剪接的上游调控网络尚未完全了解。
二、核心优势
本研究结合“多组学分析和临床前模型及临床队列“的功能验证,识别出Enza耐药性的新机制。证明KDM5B通过表观遗传抑制PTEN激活PI3K/Akt通路。这导致糖酵解酶磷酸甘油激酶1(PGK1)上调,促进代谢重编程和乳酸积累。乳酸增加后,作为p300介导的裂接因子hnRNPA1在赖氨酸179处乳化的底物。HDAC1/2可以可逆地去除这种修改。乳酸化的hnRNPA1(K179la)保持稳定,因为它避免了NEDD4L介导的泛素化和降解。该改装提升了主动AR-V7接头变体的生产效率,这是抵抗力的关键因素。研究团队还发现了一个潜在的反馈环路,即KDM5B驱动的Akt信号促进AR激活,而AR随后增加KDM5B转录。这个环路有助于维持阻力。发现表明针对KDM5B介导轴的多靶点干预可能成为克服Enza耐药性的有前景策略。
三、实验方法与主要结果
2、KDM5B通过上调PGK1重新编程葡萄糖代谢,并赋予Enza耐药性
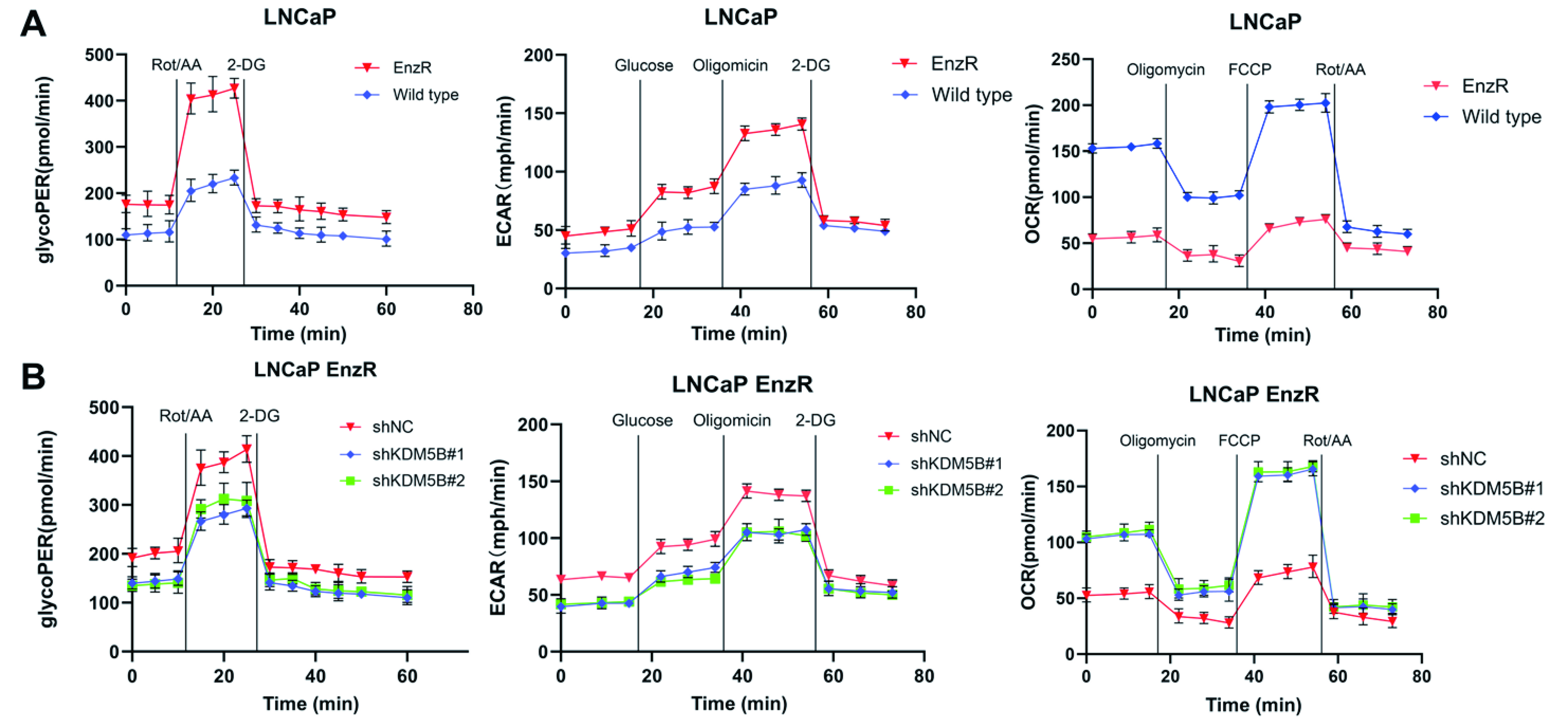
基于我们的scRNA-seq分析,显示KDM5B+耐药细胞中糖酵解通路富集。结合我们之前的数据,显示KDM5B表达与全局Pan-Kla水平呈正相关。为了验证这一点,我们首先确认了抗药细胞的代谢表型。事实上,EnzR细胞表现出高糖解状态,其特征是糖PER和ECAR相较于其敏感细胞更高。重要的是,这种糖解重编程与KDM5B的表达密切相关。这一点通过在EnzR细胞中敲低KDM5B得以证实,降低了其糖解能力、葡萄糖摄取和乳酸的产生。相反,EnzS细胞中的异位KDM5B过度表达足以增加这些糖酵解指标。因此,这些数据表明,在Enza抗性细胞中观察到的葡萄糖代谢重编程可能由KDM5B驱动。
3、KDM5B通过表观遗传机制抑制PTEN,激活PI3K/Akt通路,推动糖酵解,并促进对恩扎鲁胺的耐药性
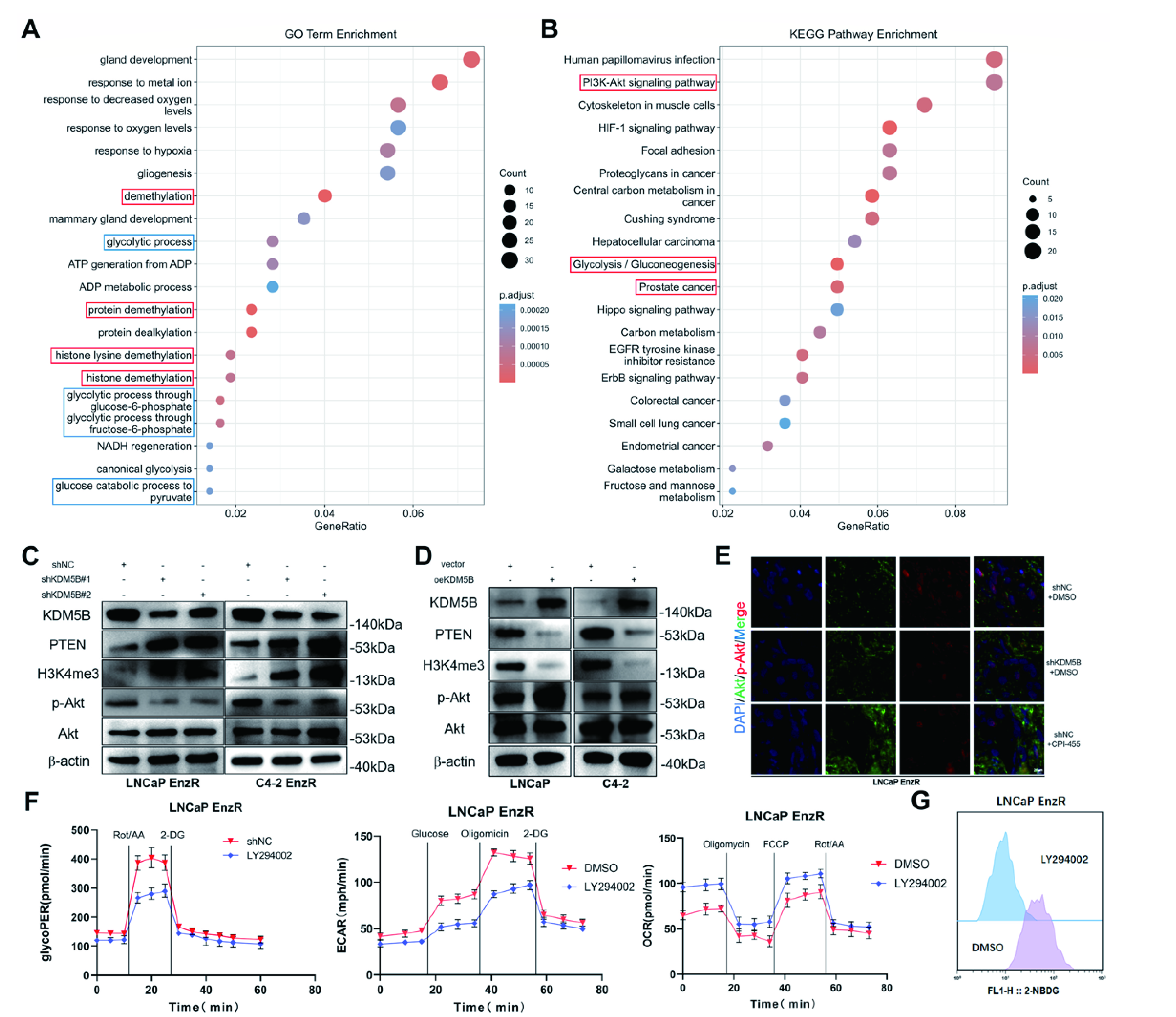
为研究KDM5B调控的下游通路,我们分析了来自KDM5B过表达RNA-seq数据集中的差异表达基因(DEGs)。从功能上讲,DEGs指向两个主要的生物学主题。首先,如预期,与KDM5B核心酶功能相关的Go术语,如“组蛋白赖氨酸脱甲基化”和“组蛋白脱甲基化”,被富集。其次,且更具机制性的是,KEGG通路分析发现了“糖酵解/糖新生”和“PI3K-Akt信号通路”的富集。这一发现与scRNA-seq分析一致,后者还显示KDM5B+耐药细胞中PI3K-Akt通路富集。
4、hnRNPA1-K179的乳化驱动AR-V7剪接,并赋予PCa中的Enza抗性
鉴于KDM5B促进糖酵解,我们首先试图将这一代谢性状与下游信号传导联系起来。TCGA-PRAD数据集的GSEA显示,高KDM5B表达组中“通过切合体进行替代mRNA剪接”通路富集(ES = 0.6876,NP = 0.0000)。这促使我们假设KDM5B驱动的乳酸积累可能影响AR切割,从而产生抗性相关变异体AR-V7。支持这一观点的是,在我们的小鼠CDX模型中,KDM5B敲低细胞的肿瘤组织乳酸水平较低。当肿瘤组织接受乳酸处理时,AR-V7 mRNA水平随时间和剂量变化增加,确立乳酸与AR-V7表达之间的直接联系。
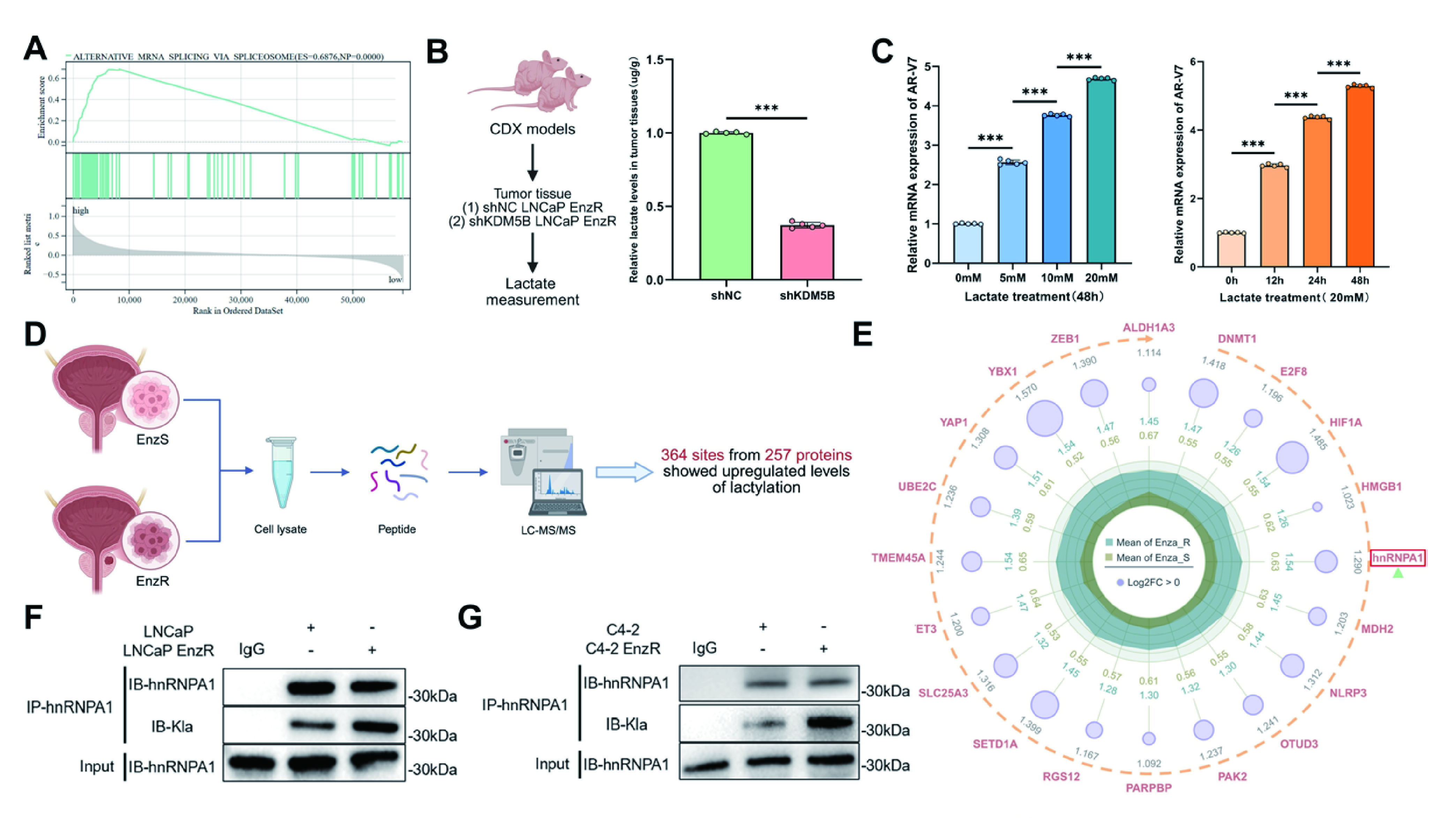
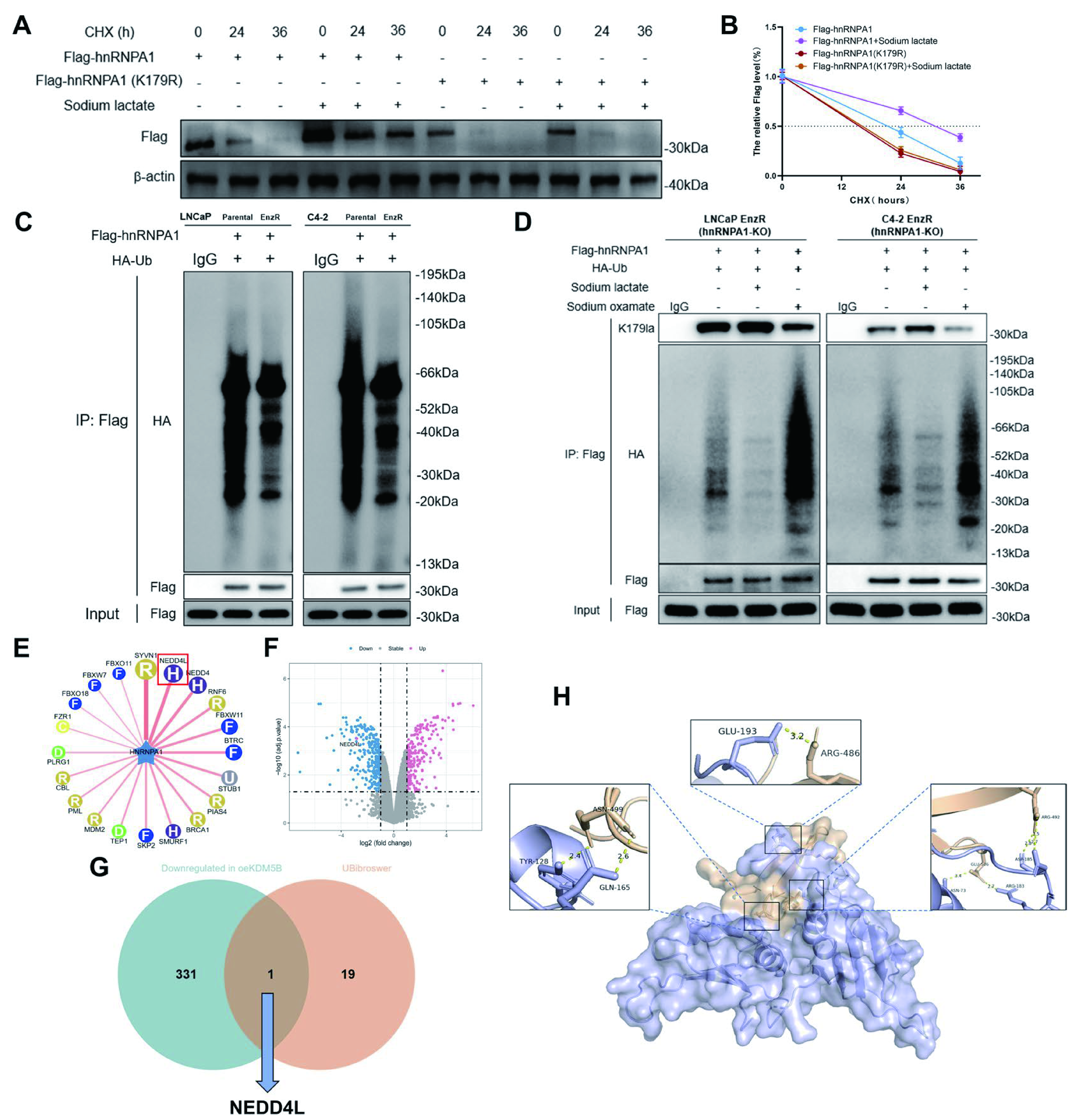
5、K179的乳化通过抑制hnRNPA1的NEDD4L介导泛素化和蛋白酶体降解来稳定其蛋白酶体
我们研究了hnRNPA1乳糖化如何改善AR-V7剪接。由于蛋白质稳定性会影响剪接因子活性,我们首先进行了环己酰胺(CHX)追踪测定。结果显示,非乳酰化的K179R突变体稳定性低于野生型(WT)hnRNPA1。此外,乳酸处理提高了WT hnRNPA1的稳定性,但未影响K179R突变体,表明K179乳酰化能增强hnRNPA1的稳定性。由于蛋白质稳定性通常受泛素化控制,我们测量了hnRNPA1泛素化水平。我们观察到EnzR细胞中的泛素化减少。与此一致的是,抑制糖酵解作用会增加hnRNPA1泛素化,而乳酸处理则产生相反效果
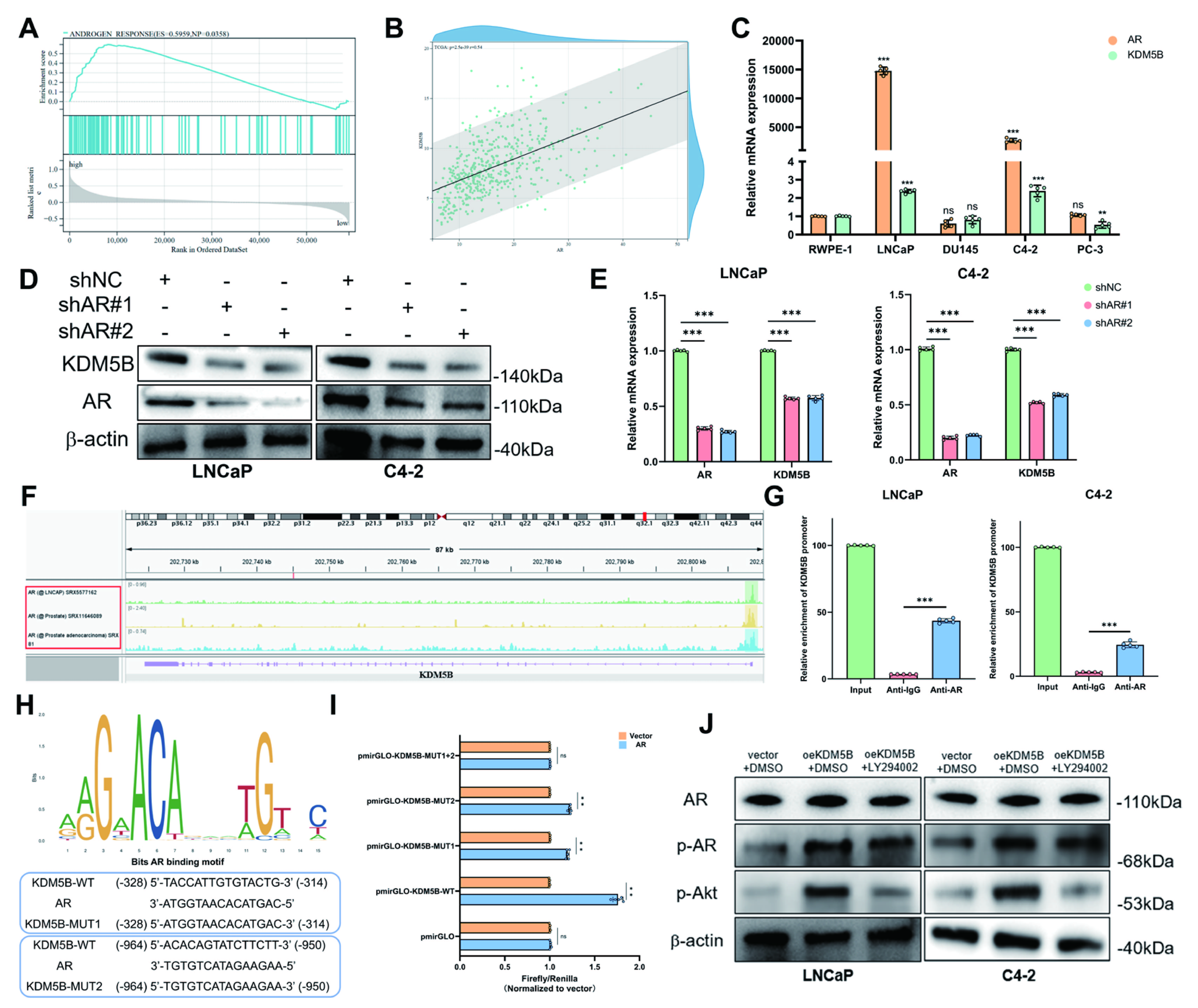
6、KDM5B与配体非依赖AR信号之间的潜在正反馈环路驱动Enza电阻
鉴于ENZA抗性PCa细胞中KDM5B表达升高,且AR在此过程中扮演重要角色,我们旨在利用TCGA-PRAD数据库中的RNA-seq谱分析KDM5B与AR之间的关系。GSEA显示KDM5B高表达组的“雄激素反应”通路显著富集,并且观察到KDM5B与AR mRNA水平之间存在强正相关。这一相关性进一步得到了我们的发现支持,即AR阳性PCa细胞系(LNCaP,C4-2)表达的KDM5B水平高于AR阴性系(DU-145,PC-3)。为了探讨潜在的直接调控关系,我们调节了AR活性。敲低AR会降低KDM5B表达,而AR过度表达则有相反效果。此外,通过AR配体二氢睾酮(DHT)刺激AR活性,或通过慢病毒递送增加AR蛋白水平,均导致KDM5B表达的剂量依赖性增加。
 四、总结
四、总结
本文研究揭示了一种新的药物耐药机制:KDM5B通过表观遗传沉默PTEN激活PI3K/Akt信号通路,进而上调PGK1,重编程肿瘤细胞中的葡萄糖代谢并促进乳酸积累。乳酸通过P300介导的hnRNPA1乳酸化增强AR-V7的表达。值得注意的是,还有一种闭环机制,涉及“KDM5B-PTEN/PI3K/Akt-AR-KDM5B”。本研究为糖酵解产生的乳酸在PCa中Enza耐药性提供了新视角。它不仅扩展了复杂的代谢、表观遗传和耐药网络,还提出了多靶点联合干预的潜在治疗策略,以逆转PCa中的Enza耐药性。
五、关于中乔新舟
本研究中RWPE-1细胞使用了RWPE-1人正常前列腺上皮细胞专用培养基(货号:ZM0351)进行培养,印证了高端科研对专用试剂产品的要求。
中乔新舟提供品种丰富的细胞培养基产品,主要分为基础培养基,细胞系完全培养基,动物原代细胞完全培养基,人原代细胞完全培养基,永生化细胞完全培养基等。该系列产品均由公司技术研发团队历经数年精心设计优化,可保持细胞良好的生长状态。所有产品均有通过严格的质控体系,经过微生物检测、PH值检测、细跑生长验证实验等确保了培养基质量的稳定性。
如果您对我们的产品感兴趣,欢迎联系我们了解更多产品详情。

研究单位:重庆医科大学发表
期刊:Molecular Cancer
影响因子:33.9
恩扎鲁胺耐药是去势抵抗型前列腺癌预后不良的重要原因,尽管已知 KDM5B 在耐恩扎鲁胺的去势抵抗型前列腺癌中高表达,但其介导耐药的具体机制尚不明确。本研究采用单细胞与多组学生物信息学分析结合体外、体内实验验证,并运用乳酰化蛋白质组学、CRISPR/Cas9、ChIP 及双荧光素酶报告实验等技术开展机制探究。结果显示,KDM5B 可通过表观遗传抑制 PTEN,激活 PI3K/Akt 通路,上调 PGK1 并引发代谢重编程与乳酸生成,进而诱导恩扎鲁胺耐药;乳酸可作为底物经 p300 介导 hnRNPA1 第 179 位赖氨酸乳酰化,阻断其 NEDD4L 介导的泛素化以稳定蛋白,促进 AR-V7 剪接,同时 KDM5B 与 AR 之间还形成正反馈环路进一步增强耐药。体内实验证实,抑制 KDM5B 或 p300 可逆转恩扎鲁胺耐药。本研究揭示了连接代谢、表观遗传与 KDM5B/AR 反馈环路的耐药新机制,提示多靶点干预有望成为克服去势抵抗型前列腺癌恩扎鲁胺耐药的有效策略。由重庆医科大学研究团队完成的成果《KDM5B-driven glucose metabolic reprogramming promotes enzalutamide resistance in prostate cancer via the lactate/hnRNPA1 lactylation/AR-V7 axis》发表于国际知名期刊《Molecular Cancer》(IF=33.9)。在该研究中,作者团队选用了中乔新舟提供的RWPE-1人正常前列腺上皮细胞专用培养基(货号:ZM0351),用于关键细胞模型的培养。
该研究确定了一种机制,将代谢、表观遗传学和KDM5B/AR反馈环与耐药性联系起来。这些发现表明,多靶点策略可能是克服CRPC中Enza耐药性的有希望的方法。
一、研究背景与科学问题
前列腺癌(PCa)仍然是全世界男性癌症相关死亡的主要原因之一。其发病率在不同地区有显著差异。雄激素受体信号抑制剂(ARSIs),如恩扎鲁胺(Enza),是晚期PCa的主要治疗方法。这些药物通过阻断雄激素受体(AR)信号来抑制肿瘤生长。然而,大多数患者最终发展为去势抵抗型前列腺癌(CRPC)对ARSIs的抗性涉及复杂的机制,其中一个关键因素是组成性活性雄激素受体剪接变异体(AR-Vs)的发展。最著名的变体AR-V7缺乏Enza所针对的配体结合域(LBD)。这种缺失使 AR-V7 持续活跃,即使在强效 ARSI 存在的情况下,也能促进亲肿瘤的转录程序。虽然AR-V7的下游效应已被充分描述,但控制其剪接的上游调控网络尚未完全了解。
二、核心优势
本研究结合“多组学分析和临床前模型及临床队列“的功能验证,识别出Enza耐药性的新机制。证明KDM5B通过表观遗传抑制PTEN激活PI3K/Akt通路。这导致糖酵解酶磷酸甘油激酶1(PGK1)上调,促进代谢重编程和乳酸积累。乳酸增加后,作为p300介导的裂接因子hnRNPA1在赖氨酸179处乳化的底物。HDAC1/2可以可逆地去除这种修改。乳酸化的hnRNPA1(K179la)保持稳定,因为它避免了NEDD4L介导的泛素化和降解。该改装提升了主动AR-V7接头变体的生产效率,这是抵抗力的关键因素。研究团队还发现了一个潜在的反馈环路,即KDM5B驱动的Akt信号促进AR激活,而AR随后增加KDM5B转录。这个环路有助于维持阻力。发现表明针对KDM5B介导轴的多靶点干预可能成为克服Enza耐药性的有前景策略。
三、实验方法与主要结果
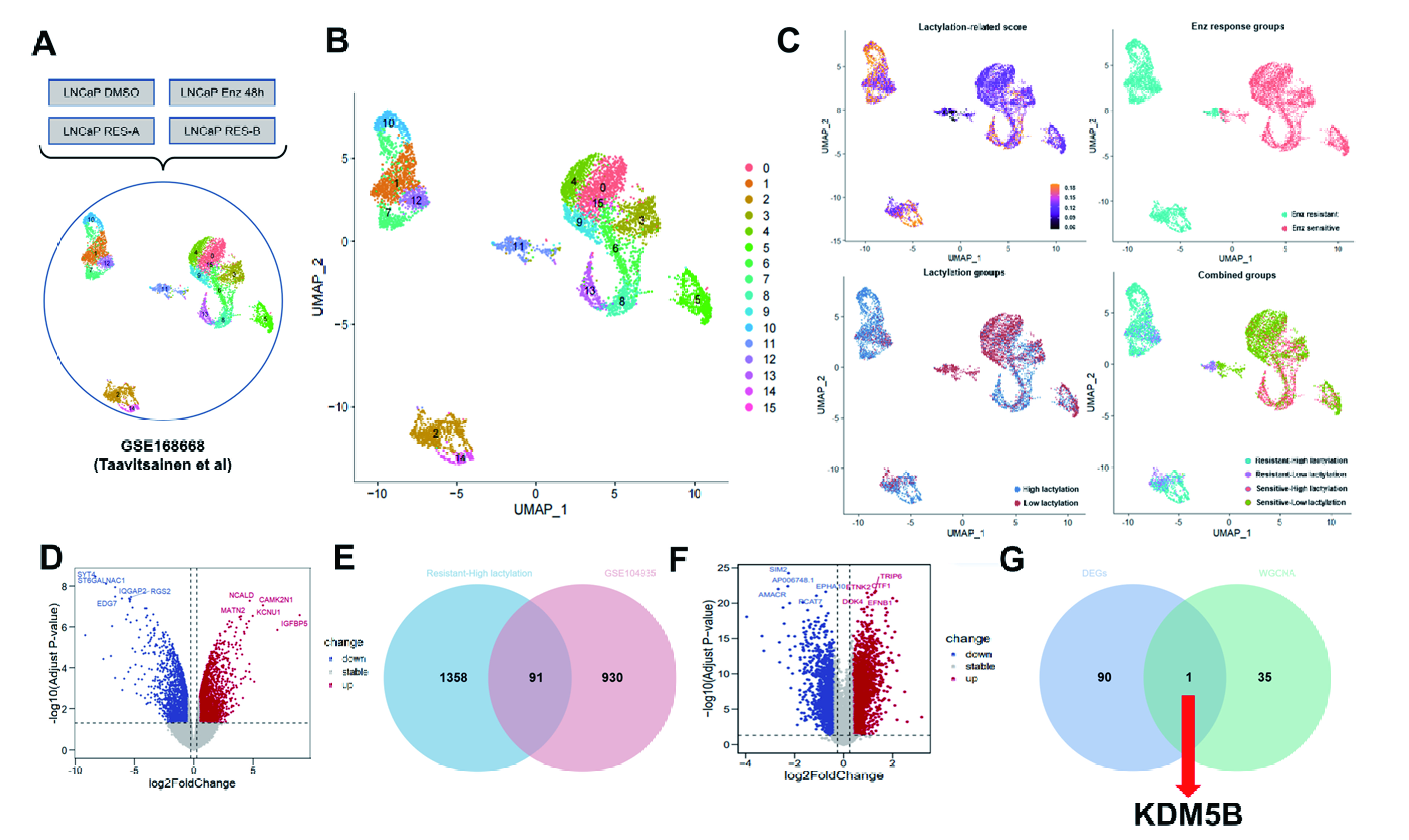
1、在高乳酸化代谢的EnzR PCa亚群中识别KDM5B
为了探讨Enza抗性的异质性,首先团队分析了亲本细胞和EnzR LNCaP细胞的scRNA-seq数据,识别出16个不同的细胞簇。通过根据细胞的耐药状态和LRG活性进行分类,确定了四个主要亚组:敏感高乳化(SH)、敏感低乳化(SL)、耐药高乳化(RH)和耐药低乳化(RL)。基因组变异分析(GSVA)显示RH簇在糖酵解和耐药相关通路方面显著富集,而CellChat分析显示该簇拥有最活跃的细胞间通信网络,在原肿瘤诱导信号(如胶原蛋白)中发挥关键作用。图1:在高乳酸化代谢的EnzR PCa亚群中识别KDM5B
A示意图,说明单细胞RNA测序(scRNA-seq)分析的工作流程。
B图均匀流形近似与投影(UMAP)图,显示了从scRNA-seq数据集中识别出的16个不同细胞簇。
C细胞群体的分层。细胞按样本来源分类(右上角为Enza敏感型与抗性型)及由“AUCell”算法推导出的乳酸相关基因(LRG)特征评分(左上角)。
Enza敏感和抗性样品之间的差异表达基因(DEGs)的火山图GSE104935数据集(|logFC| < 0.5,p< 0.05).
B图均匀流形近似与投影(UMAP)图,显示了从scRNA-seq数据集中识别出的16个不同细胞簇。
C细胞群体的分层。细胞按样本来源分类(右上角为Enza敏感型与抗性型)及由“AUCell”算法推导出的乳酸相关基因(LRG)特征评分(左上角)。
Enza敏感和抗性样品之间的差异表达基因(DEGs)的火山图GSE104935数据集(|logFC| < 0.5,p< 0.05).
2、KDM5B通过上调PGK1重新编程葡萄糖代谢,并赋予Enza耐药性
图2:KDM5B通过上调PGK1重新编程葡萄糖代谢,并赋予恩扎耐药性
海马XF代谢通量分析。比较Enza敏感(EnzS)和抗性(EnzR)LNCaP细胞(A)在KDM5B敲低后(B)及亲本细胞KDM5B过表达(C)后,在EnzR细胞中的糖解质子外流速率(GlycoPER)、细胞外酸化速率(ECAR)和氧气消耗速率(OCR)。
使用荧光葡萄糖类似物2-NBDG进行葡萄糖摄取测定。定量KDM5B敲低EnzR细胞(D)和KDM5B过度表达的亲本细胞(E)中的葡萄糖摄取。
KDM5B 敲除 EnzR 细胞(左)和 KDM5B 过表达亲代细胞(右)的乳酸产生测定。
使用荧光葡萄糖类似物2-NBDG进行葡萄糖摄取测定。定量KDM5B敲低EnzR细胞(D)和KDM5B过度表达的亲本细胞(E)中的葡萄糖摄取。
KDM5B 敲除 EnzR 细胞(左)和 KDM5B 过表达亲代细胞(右)的乳酸产生测定。
3、KDM5B通过表观遗传机制抑制PTEN,激活PI3K/Akt通路,推动糖酵解,并促进对恩扎鲁胺的耐药性
图3:KDM5B通过表观遗传机制抑制PTEN激活PI3K/Akt通路,驱动糖酵解,并促进对enzalutamide的耐药性
RNA-seq数据的基因本体(GO)和KEGG功能通路。
Western blot 分析显示,KDM5B 的敲低会增加 EnzR 细胞(C)中的 PTEN 并降低 p-Akt 水平,而 KDM5B 过表达则在亲本细胞(D)中产生相反效果。H3K4me3水平被标为KDM5B脱甲基化酶活性的对照。
免疫荧光染色显示,在不同治疗条件下,异种移植肿瘤中Akt(绿色)和p-Akt(红色)均定位。图像通过共聚焦激光扫描显微镜(比例:20微米)拍摄。
RNA-seq数据的基因本体(GO)和KEGG功能通路。
Western blot 分析显示,KDM5B 的敲低会增加 EnzR 细胞(C)中的 PTEN 并降低 p-Akt 水平,而 KDM5B 过表达则在亲本细胞(D)中产生相反效果。H3K4me3水平被标为KDM5B脱甲基化酶活性的对照。
免疫荧光染色显示,在不同治疗条件下,异种移植肿瘤中Akt(绿色)和p-Akt(红色)均定位。图像通过共聚焦激光扫描显微镜(比例:20微米)拍摄。
4、hnRNPA1-K179的乳化驱动AR-V7剪接,并赋予PCa中的Enza抗性
图4:hnRNPA1-K179的乳酸化驱动AR-V7剪接,并赋予PCa中的Enza抗性
基因集富集分析(GSEA)显示,TCGA-PRAD数据库中KDM5B高表达组中“通过切合体进行替代mRNA剪接”通路的富集。
CDX模型示意图(左)及不同干预组切除肿瘤乳酸水平的定量(右,n=5)(使用Biorender平台绘制)。
RT-qPCR分析表明,乳酸处理会以剂量(左)和时间依赖(右)方式提高异种移植组织中的AR-V7 mRNA水平。
CDXEnzR和CDX亲本组织乳酸化组学分析示意图(使用Biorender平台绘制)。
基因集富集分析(GSEA)显示,TCGA-PRAD数据库中KDM5B高表达组中“通过切合体进行替代mRNA剪接”通路的富集。
CDX模型示意图(左)及不同干预组切除肿瘤乳酸水平的定量(右,n=5)(使用Biorender平台绘制)。
RT-qPCR分析表明,乳酸处理会以剂量(左)和时间依赖(右)方式提高异种移植组织中的AR-V7 mRNA水平。
CDXEnzR和CDX亲本组织乳酸化组学分析示意图(使用Biorender平台绘制)。
5、K179的乳化通过抑制hnRNPA1的NEDD4L介导泛素化和蛋白酶体降解来稳定其蛋白酶体
图5.K179的乳酸化通过抑制hnRNPA1的NEDD4L介导泛素化和蛋白酶体降解来稳定其蛋白酶体
蛋白质稳定性分析。用WT或K179R突变hnRNPA1重构的hnRNPA1-KO EnzR细胞,使用蛋白质合成抑制剂环己酰亚胺(CHX)(40微分)处理,可加乳酸或无乳酸(20微分)。西方印迹(A)和hnRNPA1半衰期(B)定量显示乳酸稳定WT,但不稳定K179R hnRNPA1。
体内泛素化检测。Flag-hnRNPA1的泛素化在EnzR细胞中低于亲本细胞(C)。乳酸处理(20uM)减少,而氧酸钠(20uM)增加,hnRNPA1泛素化(D)。
蛋白质稳定性分析。用WT或K179R突变hnRNPA1重构的hnRNPA1-KO EnzR细胞,使用蛋白质合成抑制剂环己酰亚胺(CHX)(40微分)处理,可加乳酸或无乳酸(20微分)。西方印迹(A)和hnRNPA1半衰期(B)定量显示乳酸稳定WT,但不稳定K179R hnRNPA1。
体内泛素化检测。Flag-hnRNPA1的泛素化在EnzR细胞中低于亲本细胞(C)。乳酸处理(20uM)减少,而氧酸钠(20uM)增加,hnRNPA1泛素化(D)。
6、KDM5B与配体非依赖AR信号之间的潜在正反馈环路驱动Enza电阻
图6.KDM5B与配体非依赖的AR信号之间的正反馈环路驱动了Enza的抵抗
GSEA显示TCGA-PRAD数据库中KDM5B高表达组“雄激素反应”通路富集。
TCGA-PRAD队列(B)及多种前列腺细胞系(C)中KDM5B与AR mRNA表达的相关分析。
西方墨迹(D)和RT-qPCR(E)分析显示,AR敲低会降低KDM5B表达。
GSEA显示TCGA-PRAD数据库中KDM5B高表达组“雄激素反应”通路富集。
TCGA-PRAD队列(B)及多种前列腺细胞系(C)中KDM5B与AR mRNA表达的相关分析。
西方墨迹(D)和RT-qPCR(E)分析显示,AR敲低会降低KDM5B表达。
本文研究揭示了一种新的药物耐药机制:KDM5B通过表观遗传沉默PTEN激活PI3K/Akt信号通路,进而上调PGK1,重编程肿瘤细胞中的葡萄糖代谢并促进乳酸积累。乳酸通过P300介导的hnRNPA1乳酸化增强AR-V7的表达。值得注意的是,还有一种闭环机制,涉及“KDM5B-PTEN/PI3K/Akt-AR-KDM5B”。本研究为糖酵解产生的乳酸在PCa中Enza耐药性提供了新视角。它不仅扩展了复杂的代谢、表观遗传和耐药网络,还提出了多靶点联合干预的潜在治疗策略,以逆转PCa中的Enza耐药性。
五、关于中乔新舟
本研究中RWPE-1细胞使用了RWPE-1人正常前列腺上皮细胞专用培养基(货号:ZM0351)进行培养,印证了高端科研对专用试剂产品的要求。
中乔新舟提供品种丰富的细胞培养基产品,主要分为基础培养基,细胞系完全培养基,动物原代细胞完全培养基,人原代细胞完全培养基,永生化细胞完全培养基等。该系列产品均由公司技术研发团队历经数年精心设计优化,可保持细胞良好的生长状态。所有产品均有通过严格的质控体系,经过微生物检测、PH值检测、细跑生长验证实验等确保了培养基质量的稳定性。
如果您对我们的产品感兴趣,欢迎联系我们了解更多产品详情。
相关文章
更多 >