

利用PEAKS DIA技术深度挖掘蛋白质组“暗物质”以发现未知序列变异
2026-05-11 来源:本站 点击次数:396
数据非依赖采集 (DIA) 是一种用于鉴定传统方法常遗漏的低丰度肽段的技术。借助全新的 PEAKS DIA,研究人员能获得一套完整、高性能的DIA分析解决方案,该方案专为实现准确、灵敏的鉴定与定量而设计,且该工作流可与 Sequence Variant(序列变体)分析无缝整合,揭示传统数据库检索无法发现的新肽段。通过这种方法,研究人员能够探索蛋白质组“暗物质”。无论在生物学还是考古学应用中,PEAKS均能助力研究人员深入了解此前未被表征的蛋白质。本文将展示一套利用 PEAKS 发现新肽段和突变肽段的完整的DIA分析工作流。
此前研究表明,已发表的早期质谱数据中仍有高达 94% 的谱图未被鉴定[1]。PEAKS DIA 工作流可以利用 Sequence Variant 和 Novel Peptide 功能对这种“暗物质”谱图深入解析,从而揭示传统数据库搜索可能忽略的重要结果信息。DIA质谱技术已成为检测低丰度肽段的实用工具,与DDA相比,其灵敏度更高、重现性更好。由于存在广泛碎裂、非胰蛋白酶酶解肽段、修饰、低丰度以及缺乏标准参考数据库等挑战,许多早期质谱谱图未被有效解析[1,2]。因此导致研究人员忽略了可能提供线索的潜在氨基酸替换、新肽段和复杂翻译后修饰 (PTM),这也证明了灵敏且全面的DIA工作流的必要性和应用价值。
本研究将展示 PEAKS DIA 如何从人类考古样本中揭示序列变异和新肽段。我们使用了公开数据集 PXD062254 的部分数据来演示 PEAKS DIA 工作流的应用[2]。该数据集包含在不同条件下提取的古人类脑组织的蛋白质组学分析结果,为 PEAKS® Online 13 鉴定古代蛋白质组隐藏信息的能力提供了理想参考。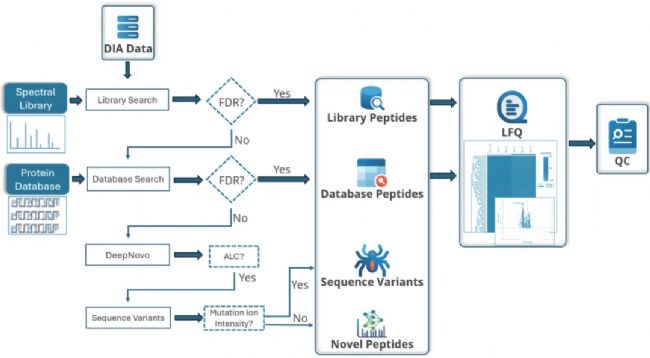
Figure 1. PEAKS DIA proteome workflow.
研究方法
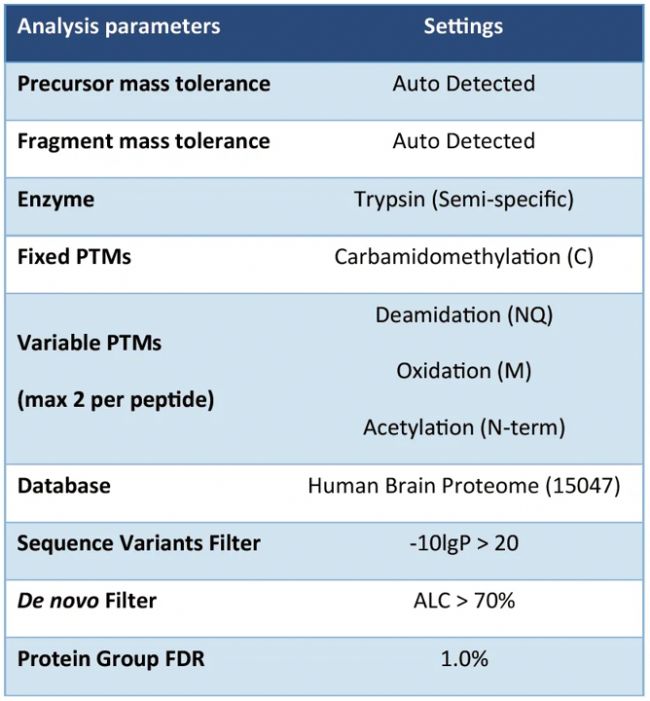
我们在PEAKS® Online 13 中使用DIA蛋白质组工作流对公开数据集[2]的数据进行检索,可自定义工作流,涵盖谱图库检索、数据库检索、序列变体分析、新肽段分析和非标记定量。分析参数见 Table 1,使用 PEAKS® Online 13 进行分析,进一步验证软件从头测序肽段检索功能,并结合 SPIDER 序列变异检索,有效挖掘古生物学样本的蛋白质组学分析结果。
Table 1. PEAKS DIA search parameters.
研究结果
01--PEAKS DIA 蛋白质组工作流
如 Figure 1 所示,PEAKS DIA 工作流整合了谱图库检索、数据库检索、序列变体检索和新肽段检索功能。允许用户根据研究需求自定义设置工作流,以探索蛋白组“暗物质”,谱图库或直接数据库搜索可单独或结合使用。
本项研究中采用了直接数据库检索的方式,所用公开数据集包含在 9 种不同条件下提取的古代脑组织样本[2]。我们使用DIA蛋白质组工作流对这些样本进行检索,旨在揭示传统数据库检索无法发现的新肽段和序列变体。
PEAKS® Online 13 是一款高通量蛋白质组学解决方案,支持多用户在项目和数据层面进行访问。它利用深度学习技术,通过算法预测保留时间 (RT)、碎片离子强度、质荷比 (m/z) 和离子迁移率,提高鉴定准确性和灵敏度,采用全新用户界面,为用户提供了一种在各类数据集上进行高质量检索的新方式。
所有样本的检索结果如 Table 2 所示。增加了 Sequence Variant 和 Novel Peptide 分析后,能鉴定更多的母离子和蛋白,所有样本总共鉴定到 8521 个 precursors,8190 条 peptides,1720 个 proteins。对比鉴定结果最高的 8 号样本,文献中鉴定到 2697 个 precursors, 2259 条 peptides 和 1157 个 protein groups,而 PEAKS® Online 13 鉴定结果显著提升,分别为 5809 个 precursors, 5628 条 peptides 和 1488 个 protein groups。
Table 2. PEAKS Online 13 DIA search results.
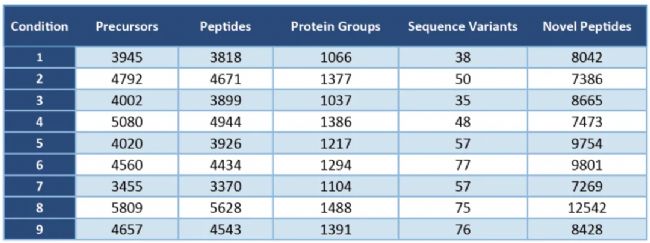
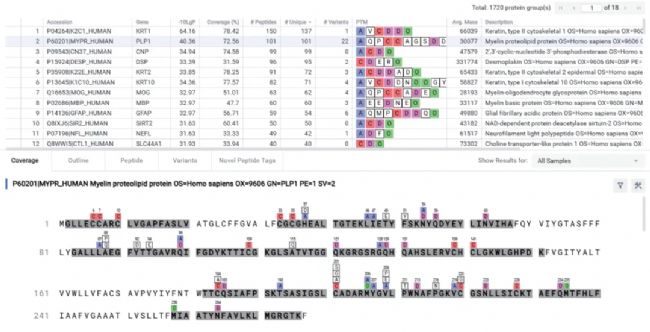
Figure 2. Table of proteins sorted by number of unique peptides and MYPR coverage map in PEAKS Online 13.
PEAKS利用基于从头测序误差与序列差异的 SPIDER 技术,来解释数据库参考序列与从头测序肽段之间的不同。 MYPR蛋白的一个已报道肽段为VG(Sub C)GSNLLSICK,突变发生在蛋白的第 220 位,由半胱氨酸(C) 突变为甘氨酸(G)。UniProt Variant Viewer 中已收录该突变,其与 Pelizaeus-Merzbacher 病密切相关,这是一种影响中枢神经系统(大脑和脊髓)疾病[3]。其他在此次分析鉴定到的存在序列变体的蛋白质还包括胶质纤维酸性蛋白 (GFAP)、Ⅰ型细胞角蛋白10 (K1C10) 和髓鞘碱性蛋白 (MBP)。
这些蛋白质均在维持大脑髓鞘结构中发挥重要作用,这也是它们在样本中相对丰度较高的原因。此外,我们还发现了以下蛋白质的已知变体:GFAP 96 位由丙氨酸(A) 替换为脯氨酸(P),对应肽段P(Sub A)LAAELNQLR;K1C10 231 位由丙氨酸(A) 替换为缬氨酸(V),对应肽段LAV(Sub A)DDFR;MBP 225 位由赖氨酸(K)替换为缬氨酸(V),对应肽段FFE(Sub K)E(Sub N)IVTPR,这些变体均已被 UniProt 收录。
检索发现了一个新肽段LLETYFSK,其MS2谱图、survey scan、precursor profile和碎片离子XIC如 Figure 3 所示,其详细匹配信息见 Figure 4。
PEAKS® Online 13 在 “Novel Peptide tab” 中提供了详细的可视化新肽段结果,方便用户选择性研究感兴趣的每个肽段。
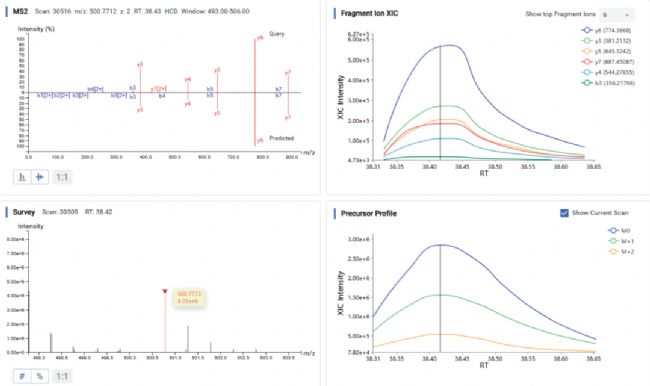
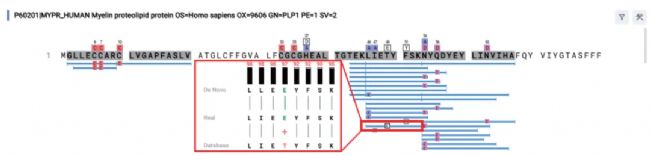
Figure 4. Alignment between de novo sequence LLEEYFSK, database and real peptide for MYPR.
03--利用非标记定量(LFQ)筛选最佳提取条件
PEAKS® Online 13 DIA 工作流还可对蛋白质进行非标记定量 (LFQ)。PEAKS LFQ 不仅能评估生物样本中肽段和蛋白质表达的变化,还能衡量不同提取条件的结果,以优化特定方法,这在分析工作流中至关重要。PEAKS DIA LFQ 基于 MS2 fragment ion profile,提供两种蛋白质定量方法:利用 MaxLFQ 算法结合全部 unique precursors 和 unique 肽段进行定量,或采用 Top 3 unique 定量法。本数据集采用了 Top 3 的方法。
Figure 5 展示了每种不同提取条件下显著性差异大于 50 的蛋白质聚类结果,可以看出 condition 8 是最佳提取条件,因为其 log2(Ratio) 最高,表明蛋白相对含量最高。
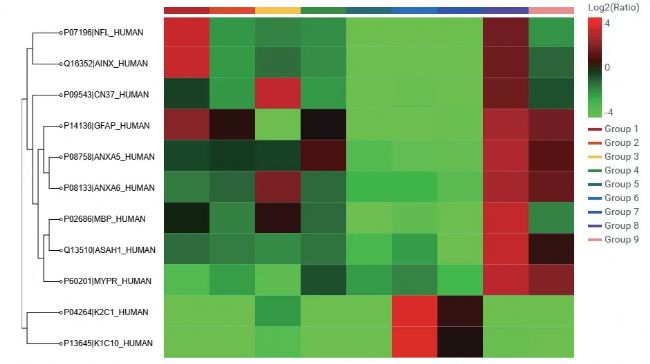
Figure 5. The result of LFQ of different sample extraction conditions.
此前研究表明,已发表的早期质谱数据中仍有高达 94% 的谱图未被鉴定[1]。PEAKS DIA 工作流可以利用 Sequence Variant 和 Novel Peptide 功能对这种“暗物质”谱图深入解析,从而揭示传统数据库搜索可能忽略的重要结果信息。DIA质谱技术已成为检测低丰度肽段的实用工具,与DDA相比,其灵敏度更高、重现性更好。由于存在广泛碎裂、非胰蛋白酶酶解肽段、修饰、低丰度以及缺乏标准参考数据库等挑战,许多早期质谱谱图未被有效解析[1,2]。因此导致研究人员忽略了可能提供线索的潜在氨基酸替换、新肽段和复杂翻译后修饰 (PTM),这也证明了灵敏且全面的DIA工作流的必要性和应用价值。
本研究将展示 PEAKS DIA 如何从人类考古样本中揭示序列变异和新肽段。我们使用了公开数据集 PXD062254 的部分数据来演示 PEAKS DIA 工作流的应用[2]。该数据集包含在不同条件下提取的古人类脑组织的蛋白质组学分析结果,为 PEAKS® Online 13 鉴定古代蛋白质组隐藏信息的能力提供了理想参考。
Figure 1. PEAKS DIA proteome workflow.
研究方法
我们在PEAKS® Online 13 中使用DIA蛋白质组工作流对公开数据集[2]的数据进行检索,可自定义工作流,涵盖谱图库检索、数据库检索、序列变体分析、新肽段分析和非标记定量。分析参数见 Table 1,使用 PEAKS® Online 13 进行分析,进一步验证软件从头测序肽段检索功能,并结合 SPIDER 序列变异检索,有效挖掘古生物学样本的蛋白质组学分析结果。
Table 1. PEAKS DIA search parameters.
研究结果
01--PEAKS DIA 蛋白质组工作流
如 Figure 1 所示,PEAKS DIA 工作流整合了谱图库检索、数据库检索、序列变体检索和新肽段检索功能。允许用户根据研究需求自定义设置工作流,以探索蛋白组“暗物质”,谱图库或直接数据库搜索可单独或结合使用。
本项研究中采用了直接数据库检索的方式,所用公开数据集包含在 9 种不同条件下提取的古代脑组织样本[2]。我们使用DIA蛋白质组工作流对这些样本进行检索,旨在揭示传统数据库检索无法发现的新肽段和序列变体。
PEAKS® Online 13 是一款高通量蛋白质组学解决方案,支持多用户在项目和数据层面进行访问。它利用深度学习技术,通过算法预测保留时间 (RT)、碎片离子强度、质荷比 (m/z) 和离子迁移率,提高鉴定准确性和灵敏度,采用全新用户界面,为用户提供了一种在各类数据集上进行高质量检索的新方式。
所有样本的检索结果如 Table 2 所示。增加了 Sequence Variant 和 Novel Peptide 分析后,能鉴定更多的母离子和蛋白,所有样本总共鉴定到 8521 个 precursors,8190 条 peptides,1720 个 proteins。对比鉴定结果最高的 8 号样本,文献中鉴定到 2697 个 precursors, 2259 条 peptides 和 1157 个 protein groups,而 PEAKS® Online 13 鉴定结果显著提升,分别为 5809 个 precursors, 5628 条 peptides 和 1488 个 protein groups。
Table 2. PEAKS Online 13 DIA search results.
02--深入探索新肽段和序列变体
筛选 PEAKS DIA Novel Peptide 分析结果中氨基酸平均置信度 (ALC) 大于 70 的肽段,序列变体分析中 -10lgP 阈值大于 20 的突变肽,并基于人 (Homo Sapiens) 蛋白质组数据库进行检索,最终发现了多种人脑相关的蛋白质(如 Figure 2 所示)。其中的髓鞘蛋白脂质蛋白 (MYPR),是中枢神经系统的主要髓鞘蛋白,在维持髓鞘结构中发挥重要作用,这与样本源自人类大脑的生物学背景相符。研究发现该蛋白质存在多个序列变体,且其序列覆盖度达 72.56%。
筛选 PEAKS DIA Novel Peptide 分析结果中氨基酸平均置信度 (ALC) 大于 70 的肽段,序列变体分析中 -10lgP 阈值大于 20 的突变肽,并基于人 (Homo Sapiens) 蛋白质组数据库进行检索,最终发现了多种人脑相关的蛋白质(如 Figure 2 所示)。其中的髓鞘蛋白脂质蛋白 (MYPR),是中枢神经系统的主要髓鞘蛋白,在维持髓鞘结构中发挥重要作用,这与样本源自人类大脑的生物学背景相符。研究发现该蛋白质存在多个序列变体,且其序列覆盖度达 72.56%。
Figure 2. Table of proteins sorted by number of unique peptides and MYPR coverage map in PEAKS Online 13.
PEAKS利用基于从头测序误差与序列差异的 SPIDER 技术,来解释数据库参考序列与从头测序肽段之间的不同。 MYPR蛋白的一个已报道肽段为VG(Sub C)GSNLLSICK,突变发生在蛋白的第 220 位,由半胱氨酸(C) 突变为甘氨酸(G)。UniProt Variant Viewer 中已收录该突变,其与 Pelizaeus-Merzbacher 病密切相关,这是一种影响中枢神经系统(大脑和脊髓)疾病[3]。其他在此次分析鉴定到的存在序列变体的蛋白质还包括胶质纤维酸性蛋白 (GFAP)、Ⅰ型细胞角蛋白10 (K1C10) 和髓鞘碱性蛋白 (MBP)。
这些蛋白质均在维持大脑髓鞘结构中发挥重要作用,这也是它们在样本中相对丰度较高的原因。此外,我们还发现了以下蛋白质的已知变体:GFAP 96 位由丙氨酸(A) 替换为脯氨酸(P),对应肽段P(Sub A)LAAELNQLR;K1C10 231 位由丙氨酸(A) 替换为缬氨酸(V),对应肽段LAV(Sub A)DDFR;MBP 225 位由赖氨酸(K)替换为缬氨酸(V),对应肽段FFE(Sub K)E(Sub N)IVTPR,这些变体均已被 UniProt 收录。
检索发现了一个新肽段LLETYFSK,其MS2谱图、survey scan、precursor profile和碎片离子XIC如 Figure 3 所示,其详细匹配信息见 Figure 4。
PEAKS® Online 13 在 “Novel Peptide tab” 中提供了详细的可视化新肽段结果,方便用户选择性研究感兴趣的每个肽段。
Figure 3. Shows the Novel Peptide LLETYFSK matched to MYPR PEAKS Online 13.
这些新肽段很多都是此前未被报道的,尤其在考古样本中(此类样本中既往突变可能未知)。此外,对该新型肽段进行BLAST分析,结果与智人样本中的髓鞘蛋白脂蛋白高度相关。
PEAKS允许用户手动查看和对比从头测序肽段与数据库参考肽段的之间的匹配结果。在 Coverage 视图中高亮显示肽段中包含的突变位点,还可以查看从头测序序列、数据库肽段与 PEAKS SPIDER 分析后的最终肽段之间的比对结果 (Figure 4)。为用户提供了一种观察潜在新突变的详细方法。
PEAKS允许用户手动查看和对比从头测序肽段与数据库参考肽段的之间的匹配结果。在 Coverage 视图中高亮显示肽段中包含的突变位点,还可以查看从头测序序列、数据库肽段与 PEAKS SPIDER 分析后的最终肽段之间的比对结果 (Figure 4)。为用户提供了一种观察潜在新突变的详细方法。
Figure 4. Alignment between de novo sequence LLEEYFSK, database and real peptide for MYPR.
03--利用非标记定量(LFQ)筛选最佳提取条件
PEAKS® Online 13 DIA 工作流还可对蛋白质进行非标记定量 (LFQ)。PEAKS LFQ 不仅能评估生物样本中肽段和蛋白质表达的变化,还能衡量不同提取条件的结果,以优化特定方法,这在分析工作流中至关重要。PEAKS DIA LFQ 基于 MS2 fragment ion profile,提供两种蛋白质定量方法:利用 MaxLFQ 算法结合全部 unique precursors 和 unique 肽段进行定量,或采用 Top 3 unique 定量法。本数据集采用了 Top 3 的方法。
Figure 5 展示了每种不同提取条件下显著性差异大于 50 的蛋白质聚类结果,可以看出 condition 8 是最佳提取条件,因为其 log2(Ratio) 最高,表明蛋白相对含量最高。
Figure 5. The result of LFQ of different sample extraction conditions.
研究结论
PEAKS DIA 工作流让用户能够以简便、友好的方式更有效地处理DIA数据。随着 PEAKS® Online 13 的全新改进,研究人员可利用 PEAKS DIA 工作流发现更多高可信度的新肽段和序列变体。经过优化的 PEAKS DIA 算法能揭示蛋白组“暗物质”,这在历史研究中具有重要意义。
参考文献
[1]Chiang Y, Welker F, Collins MJ. Spectra without stories: reporting 94% dark and unidentified ancient proteomes. Open research Europe. 2024; 4:71. https://doi.org/10.12688/openreseurope.17225.1
[2]Morton-Hayward A, Flannery S, Vendrell I, Fischer R. Deep palaeoproteomic profiling of archaeological human brains. PloSone. 2025;20(5): e0324246. https://doi.org/10.1371/journal.pone.0324246.
[3]Mimault C et al. American journal of human genetics. 1999;65(2):360–369. https://doi.org/10.1086/302483
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
PEAKS DIA 工作流让用户能够以简便、友好的方式更有效地处理DIA数据。随着 PEAKS® Online 13 的全新改进,研究人员可利用 PEAKS DIA 工作流发现更多高可信度的新肽段和序列变体。经过优化的 PEAKS DIA 算法能揭示蛋白组“暗物质”,这在历史研究中具有重要意义。
参考文献
[1]Chiang Y, Welker F, Collins MJ. Spectra without stories: reporting 94% dark and unidentified ancient proteomes. Open research Europe. 2024; 4:71. https://doi.org/10.12688/openreseurope.17225.1
[2]Morton-Hayward A, Flannery S, Vendrell I, Fischer R. Deep palaeoproteomic profiling of archaeological human brains. PloSone. 2025;20(5): e0324246. https://doi.org/10.1371/journal.pone.0324246.
[3]Mimault C et al. American journal of human genetics. 1999;65(2):360–369. https://doi.org/10.1086/302483
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
相关文章
更多 >