

文章解读:in vivo CAR-T疗法中VivoVec慢病毒递送系统的研究
2025-09-19 来源:落花微雨的知识树 点击次数:1076文章来源公众号:落花微雨的知识树 作者:心字叠衣
最近,in vivo CAR-T疗法无疑成为了生物医学领域最炙手可热的话题!从学术会议到投资论坛,从科研论文到媒体报道,这股热潮几乎席卷了整个生命科学界。网络上关于这项技术的讨论呈现出明显的两极分化——支持者视其为癌症治疗的终极解决方案,质疑者则对其安全性和商业化前景持保留态度。但无论争议如何,in vivo CAR-T疗法的迅猛发展本身就说明了其巨大的潜力和价值。
作为科研工作者,我们更需要透过现象看本质。与其被外界的喧嚣所干扰,不如静下心来深入研读几篇高质量的文献,从基础机制到临床数据,从技术瓶颈到未来方向,真正理解这项技术的核心所在。今天,就让我们一起开启这场in vivo CAR-T的文献探索之旅!
这是2024年8月份发表在blood的一篇文章:In vivo CAR T-cell generation in nonhuman primates using lentiviral vectors displaying a multidomain fusion ligand. 虽然这不是最新的文献,但是VivoVec是Umoja Biopharma开发的慢病毒递送系统,所以这篇文献还是值得学习一下的。说起来,这是解读的第二篇,第一篇详见:文献阅读[1]-CAR T cells produced in vivo to treat cardiac injury

01:文章前言
文章里作者提到了目前普通CAR-T制备的问题:一个主要的问题制备复杂,需要从每个患者身上收集大量的T细胞,将细胞运送到专门的制造中心,通过进行体外基因编辑和T细胞扩增,将其制备成最终的产品,期间需要进行QC,最终才能将CAR-T细胞产品回输。这一过程导致了在初始T细胞收集和最终产品回输之间存在大量的等待期。此外,CAR-T回输前也需要进行清淋,而且有时候伴随着较大的毒性,需要住院治疗。目前CAR-T细胞的制备和给药过程的复杂性,以及高昂的成本使得只有一小部分符合条件的患者可以获得CAR-T细胞治疗。
基于此,作者开发了VivoVec系统:
基础载体类型:是第三代自失活慢病毒载体。这类载体经过基因工程改造,删除病毒复制所需基因,且启动子区域具有自失活设计,可降低插入突变风险,符合临床安全性要求。
表面展示的功能元件:
1)载体表面展示抗CD3单链可变片段。该scFv是人工设计的抗体片段,能特异性结合T细胞表面的CD3分子,实现载体对T细胞的靶向性结合。
2)伪型化包膜蛋白:载体采用低密度脂蛋白受体趋向性的cocal融合糖蛋白进行伪型化:cocal糖蛋白是囊泡性口炎病毒G蛋白(VSV-G)的结构类似物,但具有更强的生物学特性:抗血清灭活能力,相比传统VSV-G包膜,cocal蛋白能抵抗人体补体系统的中和作用,确保载体在体内循环时保持稳定,避免被快速清除。病毒的cocal通过与细胞表面的LDLR识别,从而将CAR基因传递到T细胞内。
3)同时表达CD80和CD58共刺激配体;
02:文章正文
2.1 VVPs displaying CD80 and CD58 generate greater numbers of CAR T cells with increased in vitro and in vivo antitumor functionality.
抗原递呈细胞同时向T细胞提供peptide-MHC抗原呈递和共刺激信号,从而使得T细胞活化。这些信号对于确保T细胞激活,促进分化,获得效应功能和形成长期记忆至关重要。CD28和CD2,以及它们各自的配体CD80和CD58,是构成T细胞-抗原提呈细胞的关键共刺激信号。作者猜测,在先前展示抗CD3 scFv的VVPs中加入CD80和CD58共刺激配体,将增强其激活T细胞的能力,从而产生具有改善抗肿瘤功能的CAR T细胞。为了验证这一假设,作者构建了VVPs和共表达CD80和CD58的VVPs,并在体外培养的健康人PBMCs中评估2种共刺激配体的作用(S1A-B)。不同于典型的使用抗CD3/CD28磁珠刺激的体外制备CAR-T细胞的方案,VVPs直接添加到PBMCs中,而不需要任何外源性刺激。共刺激分子的加入大大增强了病毒颗粒结合T细胞的能力(S1C)。

而且共刺激分子的加入也导致短期T细胞活化增强,这里是VVPs加入三天后,通过流式检测CD25的表达水平验证的(1A)。

而且共刺激分子的加入也导致T细胞释放了更多的细胞因子,包括IFNγ,IL-2和TNF-α(S1D)。

不论是否含共刺激配体的VVPs都能产生相似比例的CAR-T细胞(S1E)。

但是有共刺激配体的VVPs可以产生更多的CAR-T细胞(1B)。

表型上,表达共刺激分子的VVPs产生的CAR-T细胞表面表达更多的CCR7和CD27(S1F)。这两个marker是记忆性T细胞群相关的标志物,与体外CAR-T细胞治疗的临床反应呈正相关。总的来说,这些数据表明,含有CD80和CD58共刺激配体的VVPs表现出增加的T细胞结合和激活,从而导致更多的CAR-T细胞生成,并具有较低分化的T细胞表型。

将表达或不表达共刺激分子的VVPs产生的CAR-T细胞分别与Nalm-6细胞以不同的ET比共培养,24小时后,检测细胞因子的表达。表达共刺激分子的VVPs组的CAR-T细胞比单独表达抗CD3 scFv的VVPs产生的IFNγ的量相似,但IL2和TNF-α的水平更高(1C)。

表达共刺激分子的VVPs组的CAR-T细胞与Nalm-6细胞共培养后,CAR-T细胞扩增的也更多。这里是由CellTrace dilution来判断的。从图中可以看到,Costim组的CellTrace的荧光都偏弱,说明这个组的CAR-T细胞扩增更强,造成CellTrace Dye被稀释(S1G)。

将VivoVec生成的抗CD19 CAR-T细胞(分两组:含共刺激分子CD80/CD58的VVPs vs. 仅含anti-CD3 scFv的VVPs)与表达CD19的肿瘤细胞(Nalm6)共培养。每2-4天移除旧培养基,重新添加新鲜肿瘤细胞(图中箭头表示刺激时间点),持续监测肿瘤细胞数量变化(通过荧光标记定量)。含共刺激分子的VVPs组的CAR-T细胞持续抑制肿瘤生长,即使在多次刺激后仍能有效控制肿瘤细胞数量(图中蓝色曲线维持低位)。仅含anti-CD3 scFv的VVPs组,随刺激次数增加,肿瘤细胞逐渐增殖(红色曲线上升),表明CAR-T细胞功能衰竭或持久性不足(1D)。

雌性Nod.Cg-PrkdcscidIL2rgtm1Wjl/SzJ小鼠(这个小鼠重度免疫缺陷导致T/B细胞缺失;IL2rg基因敲除缺乏NK细胞;MHC I/II双敲除)。Day -4,通过 尾静脉注射 2.5E5个 Nalm6细胞。Day -1,用 IVIS测量小鼠肿瘤负荷。根据肿瘤大小随机分组,确保组间基线一致。通过腹腔注射2E7个PBMCs。Day 0,腹腔注射携带CAR基因的 VivoVec病毒颗粒(剂量根据分组调整),体积200 μL(S1E)。

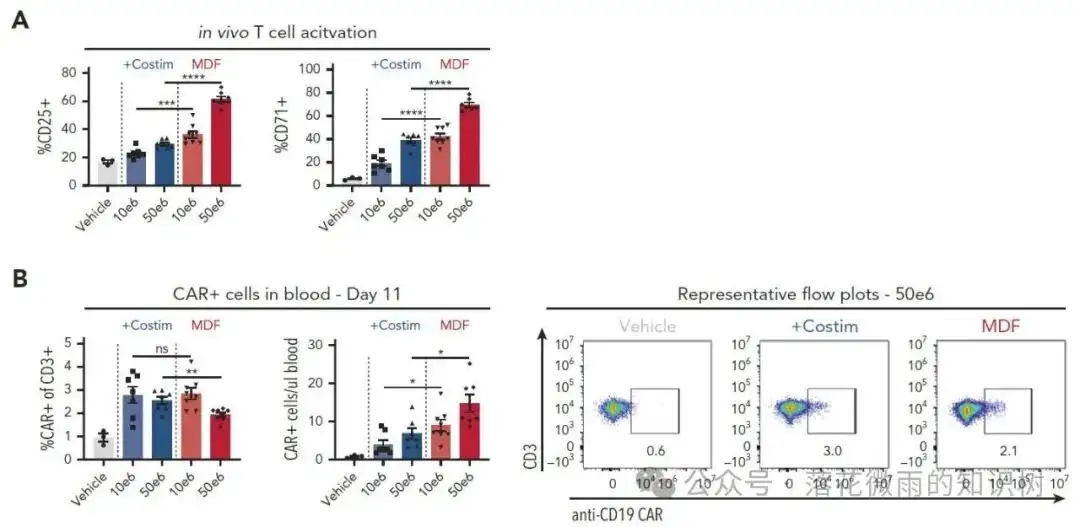
VVP给药4天后,取小鼠的外周血,通过流式细胞术定量分析人源CD3+ T细胞表面CD25和CD71的表达水平,评估其活化状态。1E的横坐标代表的是不同VivoVec病毒颗粒的注射剂量,单位为转导单位(TU)。与体外研究结果一致,表达带有共刺激配体的抗CD3 scFv的VVPs比单独表达抗CD3 scFv的VVPs诱导了更强的T细胞活化,且呈剂量依赖性。VVP给药11天后检测外周血中CAR-T细胞频率。用含共刺激配体的抗CD3 scFv的VVPs处理的动物显示出更多的CAR-T细胞数量(1F-G)。最后,对肿瘤负荷进行评估,使用共刺激配体表达抗CD3 scFv的VVPs治疗,在VivoVec病毒颗粒较低剂量下肿瘤抑制作用就很强了(1H)。

2.2 An MDF protein comprising costimulatory molecules enhances T-cell activation and transduction.
作者设计了一种包含多个结构域的蛋白(MDF):包括CD58、抗CD3 scFv和CD80组成的单链长肽。这样可以简化VVPs的制备,并且成功将MDF成功展示在病毒表面(S2A)。

出乎意料的是,与单独的抗CD3 scFv或含有共刺激分子的病毒颗粒相比,MDF VVPs与T细胞的结合要更多(2A),并导致更高的T细胞CD25表达(2B)。

但是MDF VVPs病毒颗粒诱导的细胞因子并没有明显增加(S2B)。

值得注意的是,MDF VVPs在体外能更有效地产生CAR-T细胞,尤其是在低感染复数时(2C)。

与含有共刺激分子的病毒颗粒相比,MDF VVPs制备的CAR-T细胞表面表达相当的CCR7和CD27记忆细胞标志物(S2C)。

接下来就是,与表达共刺激分子的VVPs组相比,MDF VVP产生的抗CD19 CAR-T细胞在与Nalm6细胞共培养时,CAR-T细胞扩增的量差不多(2E),IFNγ产生量差不多(2D),但产生更多的IL-2和TNF-α(2D),提示MDF VVP产生的CAR-T细胞可能具有更好的功能性。在连续刺激实验中,MDF VVP产生的CAR-T细胞能够更好地控制Nalm6肿瘤的生长(2F)。

在连续刺激实验中,MDF VVP产生的CAR-T细胞甚至优于使用传统的CD3/CD28 beads为基础的刺激方案产生的CAR-T细胞(S2D)。

2.3 MDF VVPs exhibit enhanced in vivo functionality in a humanized mouse leukemia model.
作者再次使用NSG MHC I/II KO模型验证MDF VVP的抗肿瘤作用。 VVP给药4天后,取小鼠的外周血,通过流式细胞术定量分析人源CD3+ T细胞表面CD25和CD71的表达水平,评估其活化状态。与体外研究结果一致,MDF VVPs比表达共刺激的VVPs诱导了更强的T细胞活化,且呈剂量依赖性(3A)。VVP给药11天后,作者观察到两种病毒颗粒在血液中产生了相似比例的CAR-T细胞,但MDF VVPs病毒颗粒可以产生更多的CAR-T细胞数目(3B)。这里与Figure1相比,3B中CAR-T细胞的数目明显少了很多,作者给出的解释是因为不同donor不一样的原因。
虽然两种病毒颗粒均能控制肿瘤生长,但MDF VVPs在控制肿瘤生长和总生存期方面表现略好。

这在10E6个转导单位的较低剂量时尤为明显,MDF VVPs治疗动物的中位生存期为90.5天,而表达共刺激分子的病毒颗粒组的中位生存期为62天(3D)。值得注意的是,在50E6个转导单位的较高剂量下,MDF VVP处理的动物在研究期间达到了100%的存活率。这些数据表明,在这个模型中,展示MDF蛋白的VVPs相对于表达抗CD3 scFv和表达共刺激配体的VVPs具有更好的抗肿瘤效果。

2.4 MDF VVPs administered intranodally induce potent and prolonged B-cell depletion in NHPs.
人源化小鼠模型具有局限性,并不能完全再现动物免疫系统的完整程度,特别是完全缺乏正常发育的次级淋巴组织和对异种移植物抗宿主病的易感性。为了解决这些局限性,作者在M nemestrina中开发了一个NHP模型,因为该物种允许用HIV-1衍生的慢病毒载体转导。在这个模型中,作者使用了一种NHP/人交叉反应的抗CD20 CAR,此前已经证明在恒河猴体内使用体外制造的CAR-T细胞可以消除B细胞。为了使MDF蛋白适用于该模型,作者将抗人CD3 scFv替换为抗NHP CD3 scFv。人的CD58和CD80保留下来,因为这两种蛋白在这两个物种中的序列具有高度同源性(S3A)。

评估MDF VVPs的NHP替代物对M.nemestrina CD4和CD8 T细胞的结合、激活和转导进行体外验证(S3B)。与靶细胞LLC-MK2-NLO+/-CD20共培养,验证CAR-T细胞的细胞杀伤功能(S3C)。总的来说,这些结果表明,NHP MDF VVPs结合、激活和转导NHP T细胞的能力与人MDF VVPs相当。

NHP模型:简言之,通过淋巴结注射,将携带抗CD20 CAR负载的NHP MDF VVPs以不同剂量注射到4只动物的淋巴管系统。与静脉给药途径相比,选择淋巴结给药途径可以实现有效的病毒颗粒与T细胞相互作用,同时减少全身组织对游离病毒颗粒的暴露。动物每周取血1-2次,通过流式细胞术检测病毒颗粒诱导的T细胞活化、CAR表达和随后的B细胞耗竭。同时对动物进行CRS和ICANS体征监测。在这个实验中,作者既没有清淋,也没有额外补充细胞因子。其中,1只动物(Z20020)接受了低剂量的VVPs(原剂量的五分之一)。

给药后7天,4只动物中有3只观察到NHP MDF VVP诱导的T细胞活化(4A)。在这3只动物中,NHP MDF VVP给药效果显著,在第10天产生的CAR T细胞占循环T细胞的比例高达65% (4B),相当于每微升血液中含有5963875和11182个CAR-T细胞。低剂量组的Z20020, 作者没有观察到明显的T细胞活化,CAR的表达,也没有观察到B细胞的消除。

正如预期的那样,到第7天,在所有检测到CAR-T细胞的动物中,循环B细胞都检测不到了。说明CAR-T细胞大量产生与B细胞消除是同时出现的(4C-D)。相比之下,在产生CAR-T细胞的3只动物中,循环B细胞的丢失是一致的,也非常持久。在治疗后的第56、63和76天,循环B细胞几乎检测不到(Z19051稍微差一点,B细胞在63和76还是能看到一点B细胞)。值得注意的是,1只动物( Z20106 )在第49天显示出大量的血液CAR T细胞的重新扩增,以应对在研究的第42天明显的少量B细胞的再次出现。这表明VivoVec产生的CAR-T细胞具有持久性,这预示着记忆性CAR-T细胞的产生。与记忆性T细胞的形成一致,B细胞也在第二只动物的第43天( Z19078 )的循环中重新出现,然后在第49天再次丢失。直到第63天,B细胞在该动物中仍然检测不到,这表明抗原特异性记忆CAR-T细胞功能持续存在。

在CRS方面,作者在血液(S3E)中检测到,CAR-T细胞产生的前不久就会观察到C反应蛋白(CRP)水平升高,这表明这是CAR-T细胞介导的,而不是病毒颗粒导致的。这是短暂的,随着CAR-T细胞在第14天从血液中消失,CRP水平正常化。如上所述,1只动物在第49天经历了血液CAR-T细胞的回升(4C),这也再次伴随着CRP的短暂升高。IL-6,铁蛋白和温度遵循类似的规律。除了CRS症状,作者在前2只动物中观察到轻微的震颤和短暂的癫痫。这些事件是短暂的,与第10天观察到的CAR-T细胞扩增峰值相吻合。给予托珠单抗和阿那白滞素治疗后震颤消失。作者没有观察到任何明显的肝脏毒性的迹象,与谷丙转氨酶,谷草转氨酶,碱性磷酸酶仍然保持在正常水平,除1只动物的谷丙转氨酶和谷草转氨酶在第1天出现短暂轻度升高外(S3F)。

取VVP治疗最长时间(139天)的1只动物进行剖检,20种不同组织通过ddPCR评估载体基因组整合。在注射的部位inguinal lymph node和紧接着的medial iliac lymph node可以检测到抗CD20 CAR转基因。但是在其它组织中并没有检测到,这表明在VivoVec处理后,转基因细胞主要被隔离在注射和邻近的淋巴结中几个月(4E)。

作者通过RNA ISH检测了CAR基因的生物分布(S3G)。与ddPCR数据一致,作者在注射的部位inguinal lymph node和紧接着的medial iliac lymph node可以检测到极少数细胞中CAR的表达。还在脾脏、骨髓和肺中检测到低水平的信号,作者认为这些信号是CAR+细胞向这些部位转运的标志。此外别的组织内依然检测不到。总的来说,这些数据表明,VivoVec通过淋巴内注射,在没有清淋的情况下,可以在免疫健全的NHP中有效地生成CAR-T细胞。生成的CAR-T细胞功能强大,导致B细胞清除长达76天。

03:方法参考
3.1 Vector production:
将5种质粒同时转入悬浮培养的HEK 293T细胞:gag pol(编码病毒结构蛋白(衣壳、酶等)); rev(协助病毒RNA转运出细胞核); cocal(编码低密度脂蛋白受体嗜性的病毒包膜糖蛋白(替代VSV-G)); MDF(编码多域融合蛋白); payload(携带目标CAR转基因, 如抗CD20 CAR)。转染16小时后加入含15 U/mL Denarase(一种核酸酶)的新鲜培养基,降解残留DNA/RNA以降低粘度并提高载体纯度。次日收获上清,用阴离子交换层析(Mustang Q色谱柱)初步纯化,通过切向流过滤浓缩并置换缓冲液,通过0.2 μm PES滤膜除菌过滤,最后分装冷冻最终病毒载体产品(VVPs)。
3.2 Particle binding:
PBMCs与病毒颗粒在室温下共培养1-4 h,每毫升病毒对应2E7个细胞,感染复数(也就是MOI)为2-10。使用anti-cocal antibody通过流式评估病毒颗粒/T细胞结合。这里的cocal是表达在VVPs病毒颗粒表面的糖蛋白,病毒颗粒与细胞共孵育后短时间内(1-4小时)进行的检测。这个时间点主要发生的是病毒颗粒与细胞表面的特异性结合(通过MDF蛋白),还来不及发生高效的病毒转导和细胞内基因表达。此时检测到的cocal信号几乎完全来源于附着在T细胞表面的完整病毒颗粒上的cocal包膜糖蛋白,并非来源于T细胞内部表达或产生的cocal蛋白(T细胞本身不表达cocal)。
3.3 In vitro PBMC transduction:
病毒颗粒直接加入到PBMCs中,按照每毫升对应2E6个PNMCs细胞。分别在第3天和第7天用流式检测激活和转导情况。
3.4 In vitro PBMC transduction:
取5E4个CAR-T细胞,使用CellTrace Violet荧光染料(这种染料可穿透细胞膜,与胞内蛋白质共价结合,用于追踪细胞分裂。而且细胞每分裂一次,荧光强度减半)进行标记。将标记后的CAR-T细胞与5E4个Nalm6肿瘤细胞共培养。培养体系使用X-VIVO无血清培养基(不含IL-2,以排除外源细胞因子对增殖的干扰)。5天后通过流式检测存活的CAR+细胞(通过死活染料排除死亡细胞,再通过CAR特异性抗体确认转导成功),以及通过CellTrace Violet荧光强度的稀释程度,荧光强度越低,说明细胞分裂次数越多。这样结果用于量化CAR-T细胞在肿瘤抗原刺激下的增殖能力。
3.5 PBMC-humanized mouse model:
雌性Nod.Cg-PrkdcscidIL2rgtm1Wjl/SzJ小鼠(这个小鼠重度免疫缺陷导致T/B细胞缺失;IL2rg基因敲除缺乏NK细胞;MHC I/II双敲除)。Day -4,通过 尾静脉注射 2.5E5个 Nalm6细胞。Day -1,用 IVIS测量小鼠肿瘤负荷。根据肿瘤大小随机分组,确保组间基线一致。通过腹腔注射2E7个PBMCs。Day 0,腹腔注射携带CAR基因的 VivoVec病毒颗粒(剂量根据分组调整),体积200 μL。监测指标肿瘤负荷和血液中的CAR T细胞生成及免疫细胞变化。这里作者用腹腔注射PBMC的原因是什么?腹腔内含大网膜、肠系膜淋巴结等次级淋巴器官,是免疫细胞归巢和激活的理想场所。注入腹腔的PBMCs可通过 腹膜淋巴管进入循环系统(需2-5天)。实验设计中,Day -1注射PBMCs后,Day 0给予VivoVec,预留了T细胞迁移至淋巴组织的时间。但是如果静脉注射,PBMCs可能滞留肺部或脾脏,腹腔注射则提供更稳定的局部微环境。
3.6 NHP model:
该实验将编码抗CD20 CAR的VivoVec载体颗粒(VVPs)直接注射到猪尾猕猴的淋巴结内(最多4个,每处≤1mL)。每日监测,记录动物的体温、活动状态、食欲、粪便情况和整体健康状况。实验严格遵守动物伦理规范,每日监测动物生理状态,并在特定时间点采血进行流式分析(检测CAR T细胞生成、免疫细胞表型等)、DNA检测(用于后续的生物分布检测,如ddPCR检测载体基因组)及临床生化检验(监测血常规、生化指标等,评估潜在毒性或副作用,如CRS相关指标CRP、IL-6、铁蛋白、肝酶ALT/AST等),以评估体内CAR T细胞的生成、功能、B细胞清除效果以及安全性。对出现CRS/ICANS迹象的动物,按方案使用了托珠单抗、阿那白滞素、地西泮进行治疗,其中一只动物还使用了地塞米松。
3.7 MDF sequence:
