

KLF2通过抑制内皮BMP/SMAD信号通路介导层流抗血管钙化作用
2025-10-24 来源:本站 点击次数:855KLF2 Mediates the Suppressive Effect of Laminar Flow on Vascular Calcification by Inhibiting Endothelial BMP/SMAD1/5 Signaling
Keywords: endothelial cells; vascular calcification.
血管钙化是动脉粥样硬化晚期常见的病理表现,尤其冠状动脉钙化显著增加心血管事件风险。临床影像学分析显示,钙化灶优先分布于血流紊乱的血管分叉或弯曲区域,提示血流动力学异常可能驱动钙化进程。内皮细胞作为机械力感知的核心单元,可通过剪切应力响应调控血管稳态,层流(LSS)诱导抗炎表型,而振荡剪切力(OSS)促进内皮-间质转化(EndMT)及成骨分化。然而,血流模式如何通过分子机制调控内膜钙化尚不明确。
点击了解: 仿生流体剪切应力系统
BMP/SMAD1/5信号通路是血管钙化的关键驱动因子,其激活通过促进EndMT和成骨基因表达加速矿物沉积。值得注意的是,KLF2作为层流诱导的转录因子,具有抗炎和血管保护功能,但其对BMP通路及钙化的调控作用未被揭示。既往研究提示,KLF2表达缺失与动脉粥样硬化进展相关,而其在钙化中的功能机制存在研究空白。这一矛盾现象暗示,KLF2可能通过调控机械力-BMP信号轴影响钙化进程。
点击了解: 血流剪切力刺激实验仪
基于此,香港中文大学生物医学学院在一项研究中,阐明KLF2通过抑制内皮BMP/SMAD1/5信号通路介导层流的抗钙化作用。通过整合临床数据、细胞力学模型及基因编辑技术,发现KLF2直接结合BMP4、BMPER和SMAD1启动子,抑制其转录活性,从而阻断EndMT和磷酸钙沉积。该机制不仅解释了钙化的空间分布特征,更为KLF2-BMP的治疗策略提供了理论依据,有望突破当前以侵入性手术为主的临床困境。研究成果发表于“Circulation research”期刊题为“KLF2 Mediates the Suppressive Effect of Laminar Flow on Vascular Calcification by Inhibiting Endothelial BMP/SMAD1/5 Signaling”。

首先,通过临床观察发现冠状动脉钙化倾向发生于血流模式受干扰的动脉分支点,提示血流动力学可能决定钙化分布。为验证这一假说,研究人员招募48例患者行冠状动脉计算机断层扫描血管造影评估。聚焦左冠状动脉系统,对比分析分叉与非分叉部位钙化程度(图1A)。统计分析明确显示,钙化在分叉部位的累积显著高于非分叉区域(图1B-1F)。进一步按钙化评分分组后,所有亚组均一致显示分叉区钙化评分显著更高。这些结果证实血管钙化在患者中存在位点特异性分布模式,且与血流紊乱密切相关。
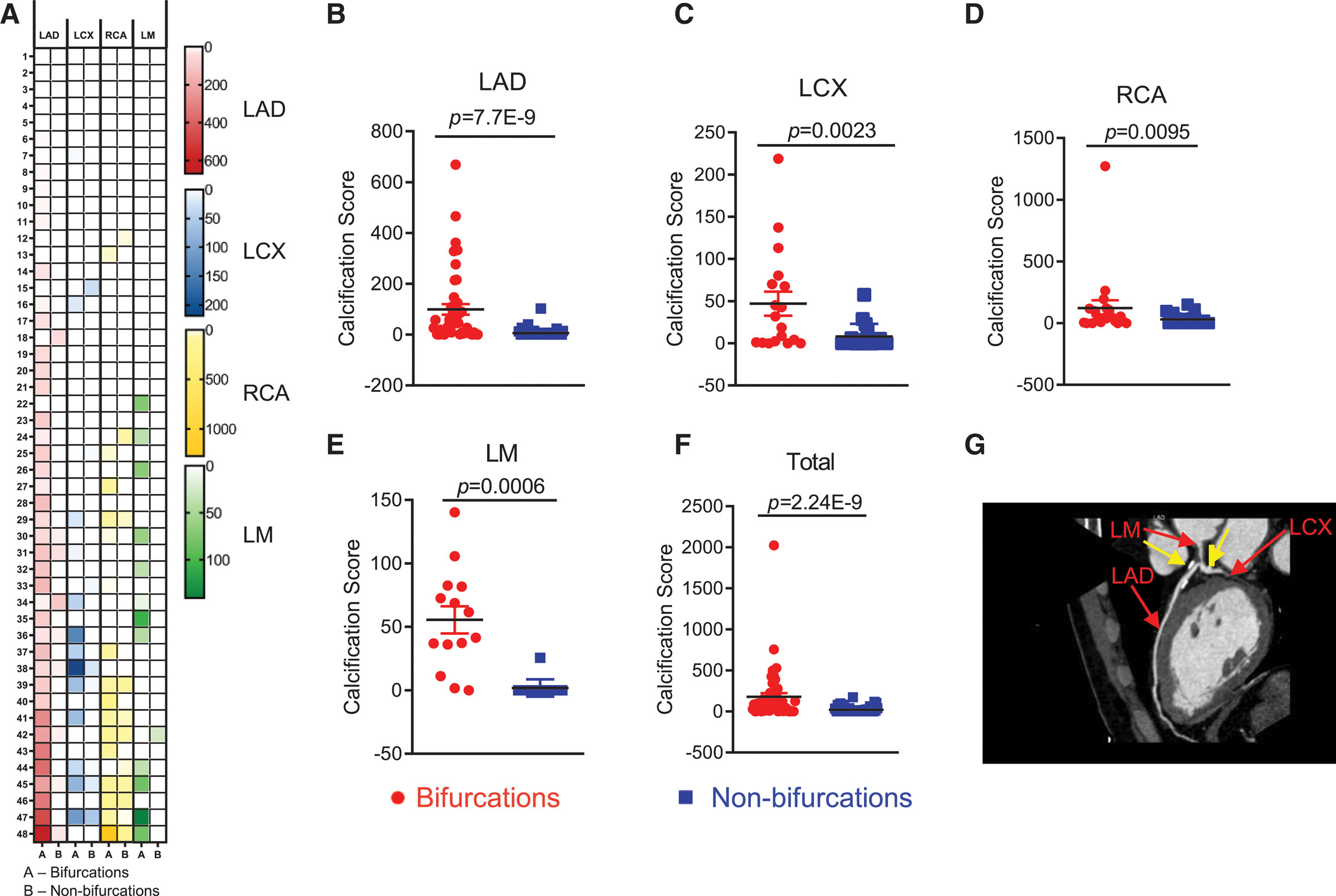
图1 患者冠状动脉钙化的部位特异性分布。
然后,由于血流动力学特征与血管钙化的空间分布密切相关,层流剪切应力(LSS)抑制钙化,而紊乱流(DF)存在于主动脉弓及分支起始部位,与钙化显著相关。为验证这一关联,研究人员通过西方饮食喂养ApoE⁻/⁻小鼠20周诱导钙化模型,骨480荧光成像显示主动脉弓钙化荧光强度显著高于降主动脉(图2A)。钙化优先发生于分支点(腹主动脉、颈动脉)及主动脉弓内曲率侧(图2B-2C),而低剪切应力的胸主动脉肋间分支点也出现钙沉积。类似现象在维生素D3过载介导的钙化模型中重现,提示血流模式对钙化分布的调控具有普适性。体外实验中,人脐静脉内皮细胞(HUVEC)在Y型流动腔室(直段20 dyne/cm²均匀LSS,分叉区10 dyne/cm²非均匀剪切应力)培养显示,分叉区钙沉积显著,而直段几乎无钙化(图2D-2E)。这表明非均匀剪切应力促进钙化,而均匀LSS通过抑制内皮细胞矿化发挥保护作用。

图2 均匀层流剪切力可抵御血管钙化。
临床分析显示,钙化性主动脉瓣狭窄患者的瓣膜钙化灶主要分布于心室侧(此处血流呈单向搏动性低剪切应力),而主动脉侧(无明确流向)钙化较少,提示局部血流动力学特征与钙化相关。进一步研究发现,钙化区域的内皮细胞KLF2表达显著低于非钙化区(图3A-3B),且血管钙化模型中KLF2水平普遍下调(图3C)。机制上,KLF2作为剪切应力敏感转录因子,其表达受血流模式直接调控,在12 dyn/cm²层流剪切应力(LSS)作用下,人脐静脉内皮细胞(HUVEC)的KLF2 mRNA在3小时内快速诱导并持续高表达至24小时(图3D);而振荡剪切应力(OSS, 0.5±4 dyn/cm²)则显著抑制KLF2表达,大鼠主动脉内皮细胞实验同样证实LSS组的KLF2水平高于OSS组(图3E)。这表明KLF2是血流动力学调控钙化的关键介质。

图3 流动敏感性的KLF2(Krüppel样因子2)在心血管钙化中被抑制。
接着,研究人员发现KLF2沉默通过诱导内皮-间质转化(EndMT)显著加剧成骨分化和血管钙化。实验显示,KLF2基因敲低导致人脐静脉内皮细胞(HUVEC)和人主动脉内皮细胞(HAEC)发生形态转变,呈现成纤维细胞样特征。基因表达分析进一步证实内皮标志物显著下调,而平滑肌细胞标志物、间质标记物及EndMT关键转录因子SNAI1显著上调,蛋白质印迹验证了上述变化(图4A)。在成骨培养基(OM)中,KLF2沉默的HUVEC表现出成骨分化标志物的异常高表达(图4B),同时骨素680荧光染色和茜素红染色显示钙沉积量显著增加(图4C-4F)。这些结果表明,KLF2缺失通过激活EndMT通路使内皮细胞获得间充质/成骨表型,直接驱动血管钙化进程,揭示了KLF2在抑制病理性钙化中的核心作用。
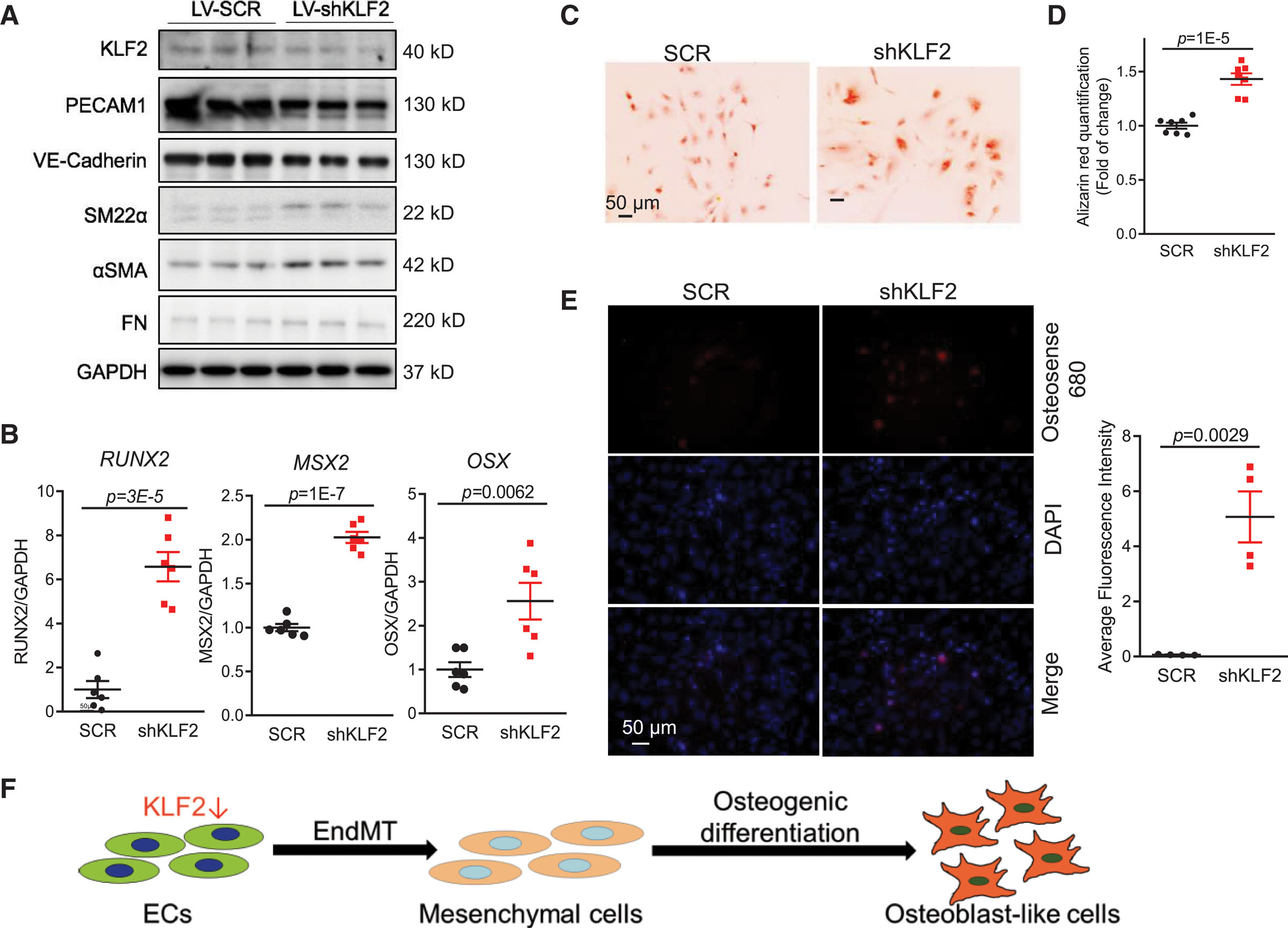
图4 KLF2(Krüppel样因子2)的沉默促进了内皮-间质转化,并加速了内皮细胞中的成骨诱导。
为明确内皮KLF2在血管钙化中的作用,研究人员构建了ApoE⁻/⁻小鼠内皮特异性KLF2敲低模型(KLF2ᴱᶜᴷᴰ)。通过尾静脉注射靶向EC的AAV9至Cas9ᶠˡ/ᶠˡ ApoE⁻/⁻和CDH5-Cre Cas9ᶠˡ/ᶠˡ ApoE⁻/⁻小鼠(图5A),成功实现内皮KLF2下调(图5B-5C),且内皮功能标志物NOS3同步下调。Western饮食喂养18周后,KLF2ᴱᶜᴷᴰ组小鼠代谢参数与对照组无差异,但近红外钙示踪显示血管钙化程度显著增加(图5D-5E)。主动脉钙含量测定进一步证实KLF2ᴱᶜᴷᴰ组钙沉积升高(图5F),并伴随成骨标志物表达上调(图5G)。这些结果证实内皮KLF2缺失直接促进血管钙化,为临床中血流紊乱区域钙化加剧提供了分子解释。
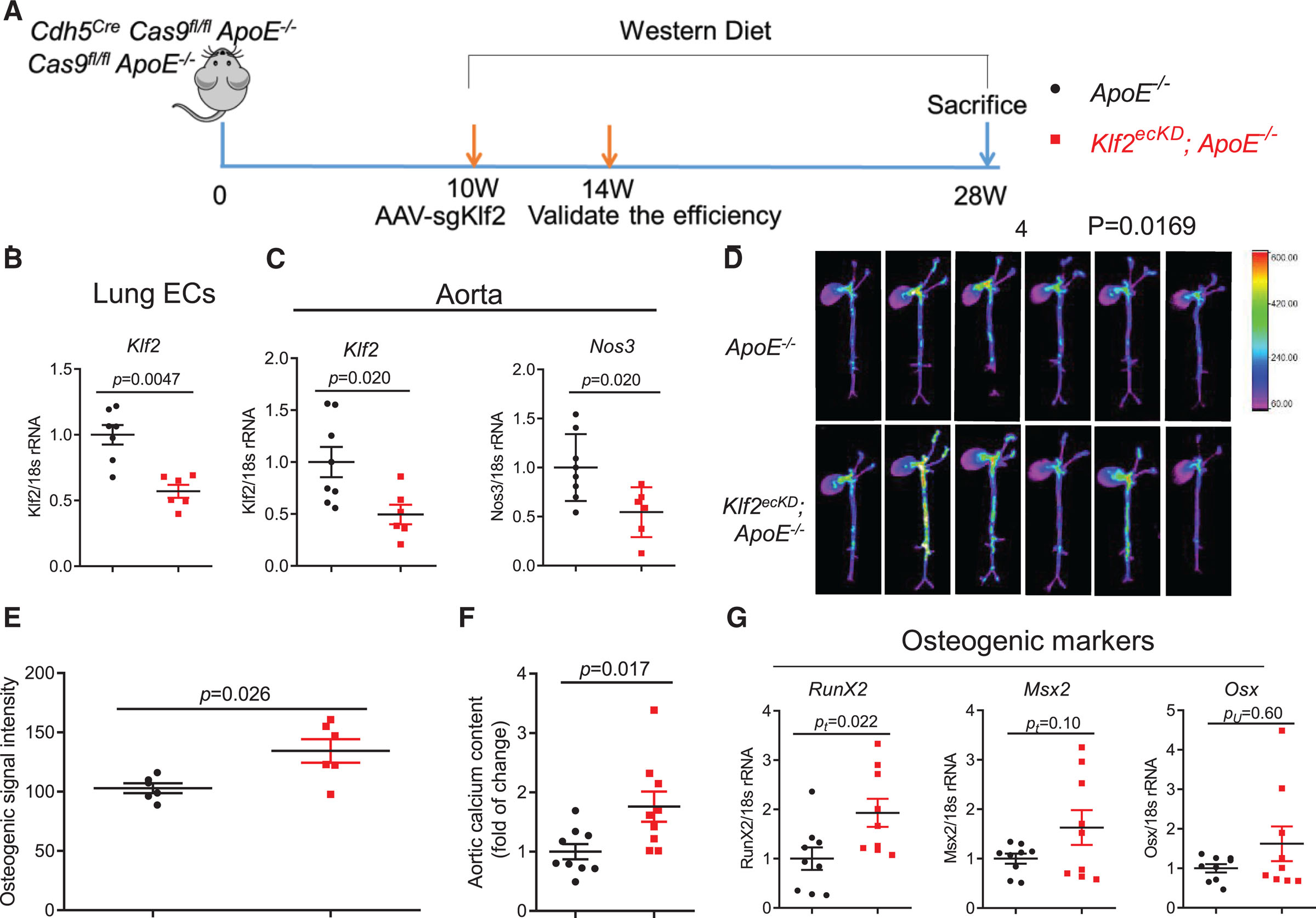
图5 内皮细胞特异性KLF2(Krüppel样因子2)基因敲低,可增加ApoE⁻/⁻小鼠的血管钙化。
接下来,为验证KLF2作为血管钙化治疗靶点的潜力,研究人员通过AAV9载体介导内皮特异性过表达干预ApoE⁻/⁻小鼠(图6A)。Western饮食喂养18周后,AAV-CDH5-KLF2组小鼠呈现以下显著变化,近红外钙示踪显示血管钙化区域减少(图6B-6C),主动脉钙含量显著下降(图6D);主动脉KLF2及其下游保护因子NOS3表达上调(图6E);成骨标志物表达下调(图6F)。这些结果证明内皮特异性KLF2过表达可有效改善血管钙化,为靶向KLF2的基因治疗策略提供了临床前证据。
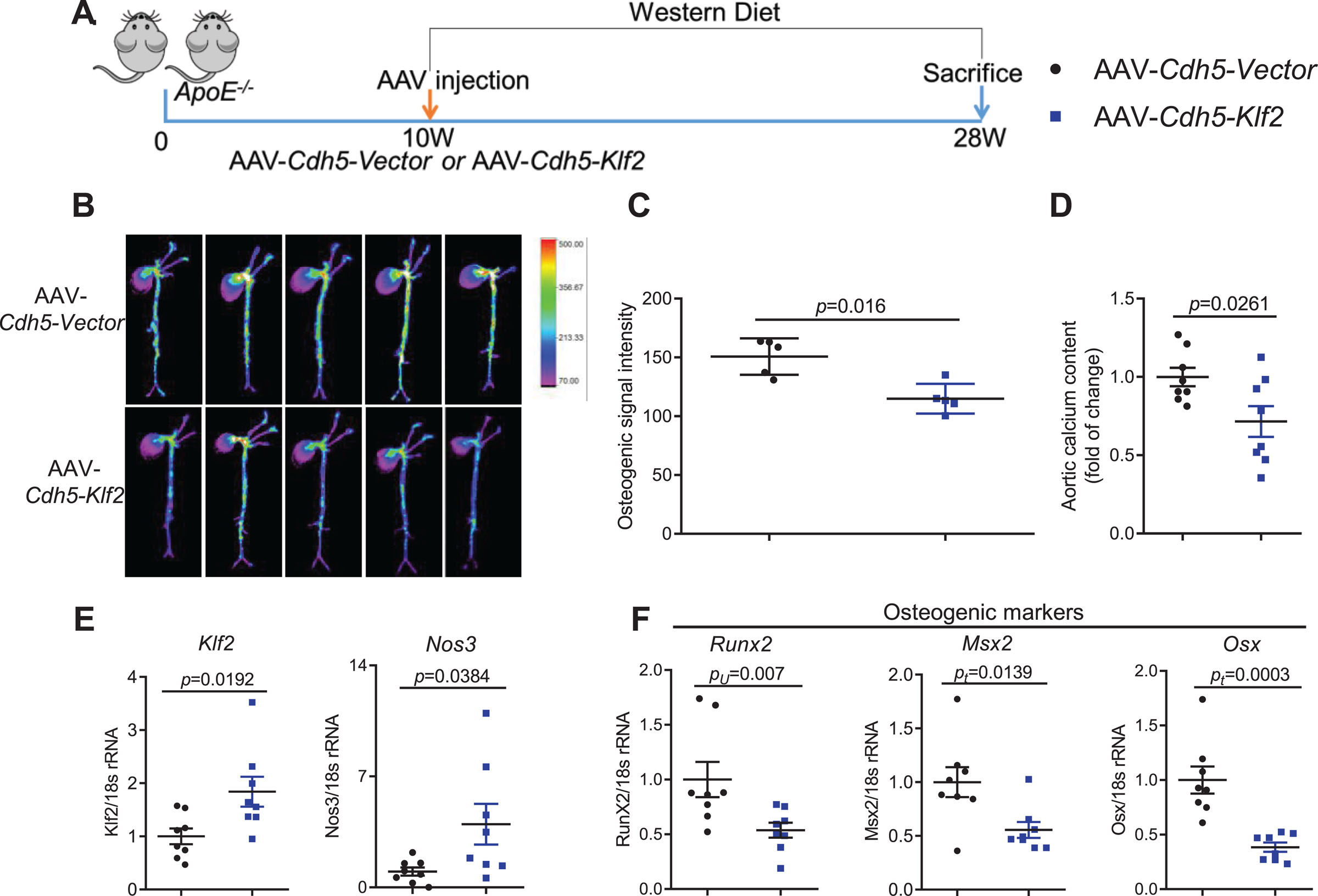
图6 利用腺相关病毒介导的、靶向内皮细胞的KLF2(Krüppel样因子2)过表达,能够改善ApoE基因敲除小鼠的血管钙化。
进一步研究发现,KLF2通过直接调控BMP配体与拮抗剂表达抑制SMAD1/5信号通路。mRNA谱分析显示,KLF2过表达显著下调BMP2/4/6等促钙化配体,同时上调BMP拮抗剂BMPER(图7B)。相反,KLF2敲低则增加BMP4/6并抑制BMPER表达(图7C)。层流剪切应力(LSS)通过KLF2依赖性机制抑制BMP4并激活BMPER,在体外,LSS处理的HUVEC中BMP4表达降低而BMPER升高(图7D-E);在ApoE⁻/⁻小鼠模型中,内皮特异性KLF2过表达同样降低主动脉BMP4并上调BMPER(图7G-H)。机制上,染色质免疫沉淀证实KLF2直接结合BMP4启动子及BMPER启动子(图7K-L),通过转录抑制BMP4和激活BMPER双重作用阻断SMAD1/5磷酸化(图7I-J)。该发现揭示了KLF2作为机械力-表观遗传枢纽调控血管钙化的精确分子路径。
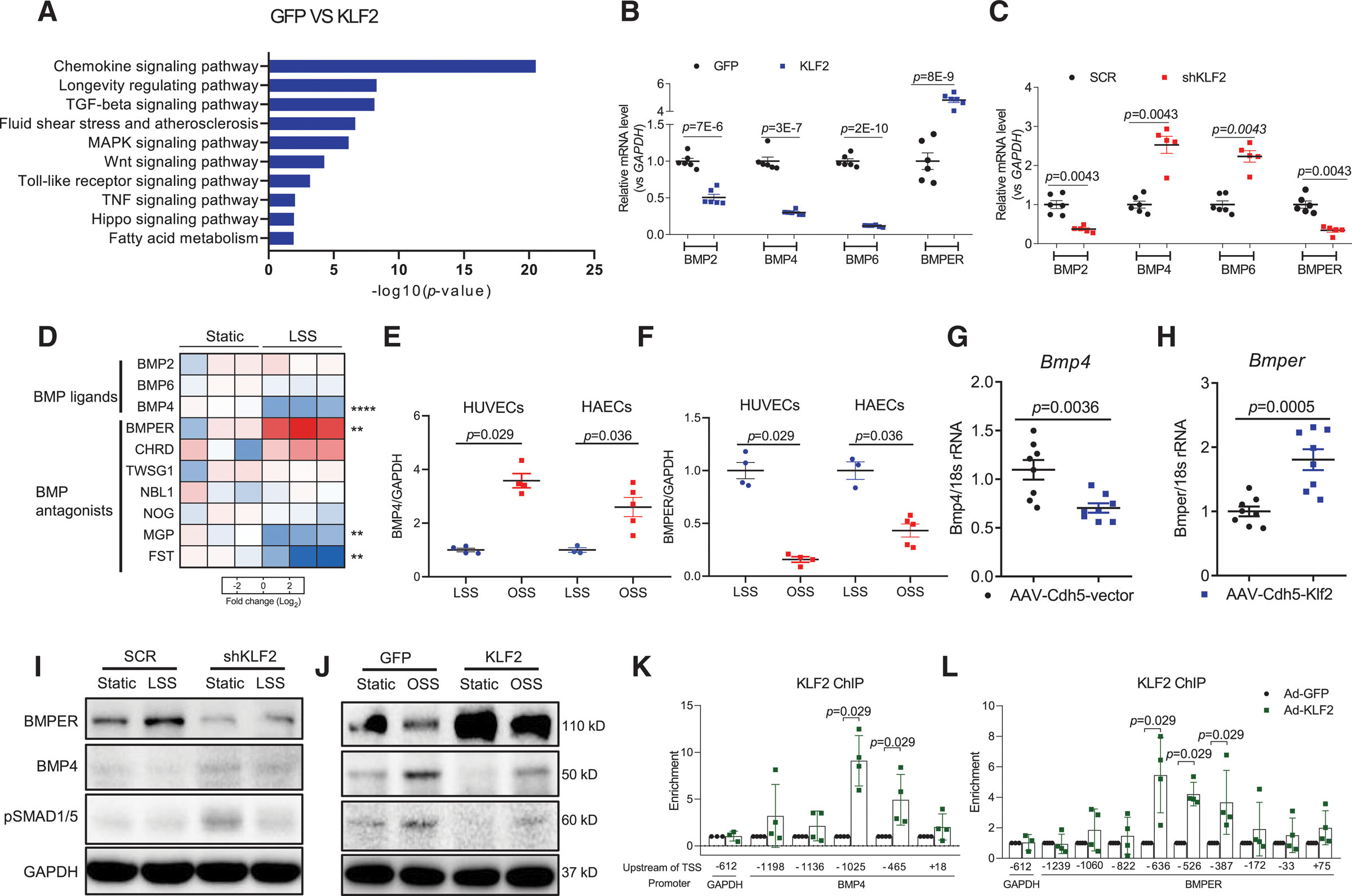
图7 KLF2(Krüppel样因子2)通过调控BMP4和BMPER,从而抑制BMP(骨形态发生蛋白)/SMAD1/5信号通路的激活。
最后,研究人员分析KLF2通过多层级调控特异性抑制SMAD1信号通路,RNA测序、qPCR及Western blot证实,KLF2过表达选择性下调人脐静脉内皮细胞(HUVEC)和人主动脉内皮细胞(HAEC)中SMAD1的表达(图8A-B)。机制上,染色质免疫沉淀显示KLF2直接结合SMAD1启动子(图8C),降低其转录活性;同时减少SMAD1核转位及染色质结合。KLF2敲低则显著升高SMAD1、p-SMAD1/5水平(图8D-E),其效应与成骨培养基(OM)处理相当(图8F)。功能验证表明,SMAD1基因沉默可wanquan逆转KLF2缺失诱导的成骨标志物表达(图8G-I),证明SMAD1是KLF2调控内皮成骨分化的核心效应因子。

图8 SMAD1介导了由KLF2(Krüppel样因子2)沉默所诱导的内皮细胞成骨分化。
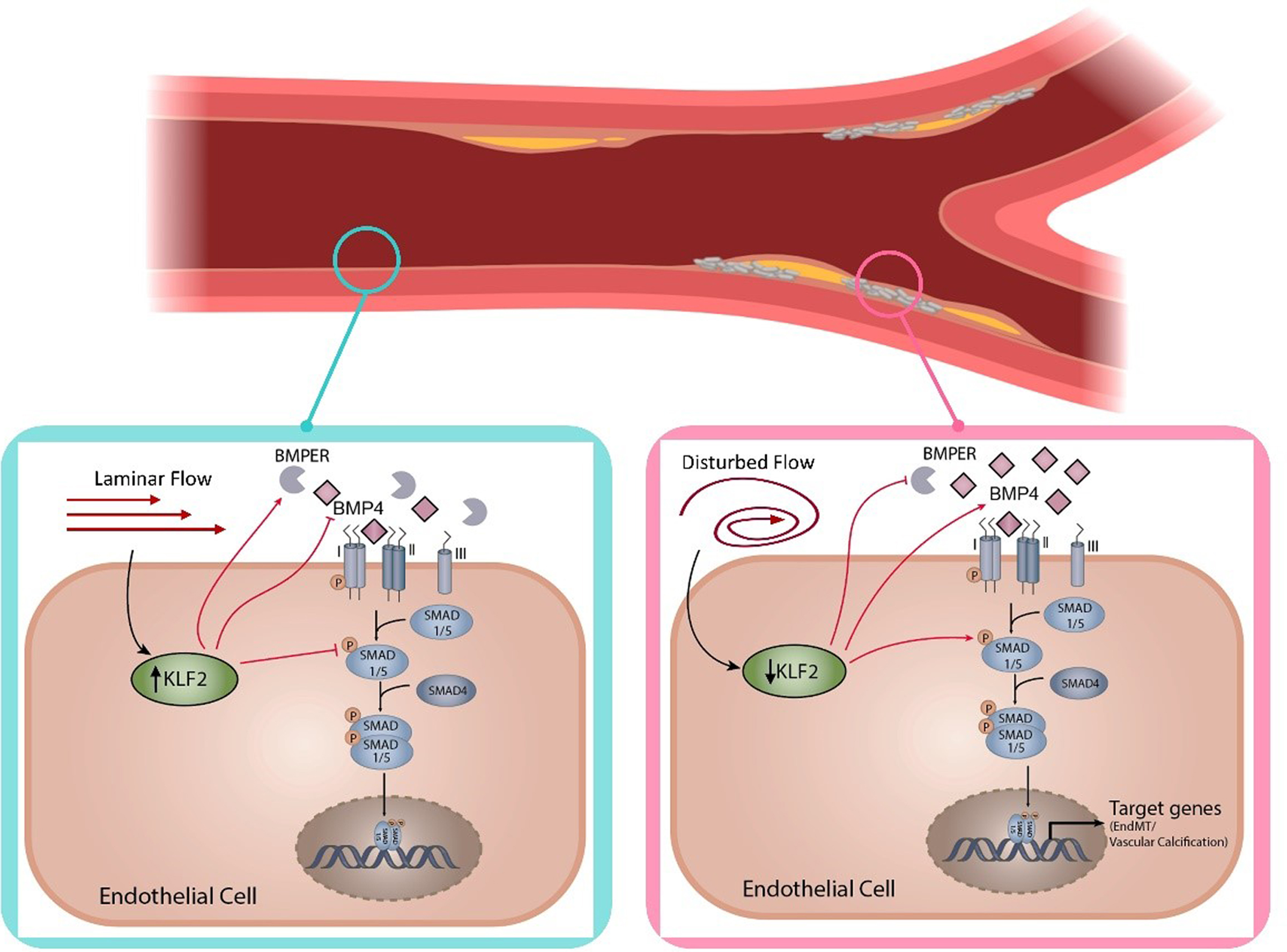
图9 图形摘要。
总之,研究揭示了KLF2通过抑制BMP/SMAD1/5信号通路介导层流剪切应力(LSS)对血管钙化的保护作用。临床数据与动物实验一致表明,钙化灶优先分布于血流紊乱区域,而内皮特异性KLF2敲除加重钙化,过表达则显著缓解。机制上,KLF2直接结合BMP4、BMPER及SMAD1启动子,抑制促钙化配体表达并激活拮抗剂。然而,研究存在局限性,瓣膜钙化中血流紊乱(DF)的具体贡献仍需量化;KLF2在成骨细胞中表现促分化功能,与内皮细胞的抑制作用相反,其细胞类型特异性机制有待解析;血管平滑肌细胞分泌的BMP2可能协同促进钙化,需进一步探究KLF2-BMP轴的多细胞互作。综上,KLF2-BMP/SMAD1/5通路为靶向血管钙化提供了新方向,但其跨细胞调控网络及临床转化潜力需深入验证。
参考文献:Huang J, Pu Y, Zhang H, Xie L, He L, Zhang CL, Cheng CK, Huo Y, Wan S, Chen S, Huang Y, Lau CW, Wang L, Xia Y, Huang Y, Luo JY. KLF2 Mediates the Suppressive Effect of Laminar Flow on Vascular Calcification by Inhibiting Endothelial BMP/SMAD1/5 Signaling. Circ Res. 2021 Aug 6;129(4):e87-e100. doi: 10.1161/CIRCRESAHA.120.318690. Epub 2021 Jun 23. PMID: 34157851.
原文链接:https://pubmed.ncbi.nlm.nih.gov/34157851/
图片来源:所有图片均来源于参考文献
小编旨在分享、学习、交流生物科学等领域的研究进展。如有侵权或引文不当请联系小编修正。如有任何的想法以及建议,欢迎联系小编。感谢各位的浏览以及关注!进入官网www.naturethink.com或关注“Naturethink”公众号,了解更多相关内容。
点击了解: 仿生流体剪切应力系统
点击了解: 血流剪切力刺激实验仪