

InnoScan 文献解读:CBL功能缺失突变掲示人类B细胞免疫核心机制
2026-04-07 来源:本站 点击次数:149
引言
在免疫系统的复杂调控网络中,E3泛素连接酶CBL(Casitas B-lineage lymphoma)一直被认为是T细胞发育和功能的关键调节因子。此前基于小鼠模型的研究明确指出,CBL对T细胞受体信号具有重要的负调控作用,其缺失会导致T细胞发育异常和功能亢进。然而,CBL在人类免疫系统中的真实角色,特别是其对B细胞的影响,长期以来都未能得到直接证实,因为小鼠研究显示CBL单独缺失并不影响B细胞功能,其作用被其同源蛋白CBL-B所冗余替代。
2026年1月15日发表于《Nature Immunology》的一项突破性研究,彻底颠覆了这一传统认知。由Stuart G. Tangye和Jonathan Bohlen领导的国际联合研究团队,通过对一群携带体细胞CBL功能缺失突变的罕见患者进行长达数年的深入分析,揭示了CBL在人类免疫系统中一个此前被完全忽略的核心作用。与小鼠模型截然相反,人类CBL对T细胞的功能影响甚微,但对于B细胞的发育、成熟、耐受性建立以及体液免疫记忆的形成而言,CBL却是不可或缺的。该研究不仅首次在人体内证实了CBL在B细胞谱系中的非冗余功能,还为理解某些原发性免疫缺陷病和自身免疫病的发病机制提供了全新的视角,并提出了潜在的治疗新策略。
方法
本研究是一项结合了临床队列分析、多组学技术、体外细胞模型和基因编辑技术的综合性研究。研究团队建立了一个由来自9个家庭的11名具有CBL杂合性缺失(LOH)的个体组成的独特队列,此外还包括8名携带遗传性杂合CBL UbLOF变异的个体(包括家庭成员及无亲缘关系者)。为了深入探究CBL缺失对B细胞发育的细胞自主性影响,研究团队运用了先进的基因编辑技术。他们从健康脐带血中分离出CD34+造血干/祖细胞(HSPCs),并使用CRISPR-Cas9系统对其进行编辑,构建了与患者基因型一致的CBL功能缺失突变模型。通过体外诱导分化体系,将这些编辑后的HSPCs向B细胞定向分化,从而在等基因背景下直接观察CBL缺失对早期B细胞发育的直接影响。此外,研究还通过腺嘌呤碱基编辑技术构建了PIK3CD功能获得性突变细胞作为对照,并通过RNA测序比较了两者在转录组水平上的异同。
在B细胞功能研究方面,研究者使用了多种精细的分析手段。通过质谱流式细胞技术对患者外周血中B细胞亚群进行了高维度的免疫表型分析。为了评估B细胞对特定抗原的反应能力,研究人员利用生物素标记的SARS-CoV-2刺突蛋白与链霉亲和素-荧光染料结合,制备了四聚体,用于检测和计数疫苗接种后外周血中抗原特异性B细胞的数量及其类别转换情况。同时,通过流式细胞术分选出不同发育阶段的B细胞亚群(过渡性B细胞、初始B细胞、记忆B细胞),并在体外给予不同刺激(如CD40L/IL-21、BCR交联、CpG等),通过酶联免疫吸附试验(ELISA)检测其分泌免疫球蛋白的能力。
尤为关键的是,本研究利用生物芯片技术对患者血浆中的自身抗体谱进行了高通量筛选。研究人员采用了HuProt人类蛋白质组芯片,该芯片上固定了超过20,000种人类重组蛋白。将患者血清与芯片孵育后,使用Alexa Fluor 647标记的抗人IgG抗体进行检测,随后通过InnoScan 1100AL荧光扫描仪对芯片进行高分辨率扫描。InnoScan 1100AL凭借其高灵敏度的光电倍增管和精确的扫描机制,能够捕捉到微弱的荧光信号,并将其转化为定量的数据。GenePix Pro软件被用于分析图像,提取每个蛋白点的信号强度,从而实现对患者血浆中针对人类自身抗原的IgG抗体进行系统性、高通量的定性及半定量分析。这一技术平台使得研究者能够全面描绘CBL缺陷患者体内自身免疫反应的广谱特征,为揭示其自身免疫表型提供了关键证据。

结果
研究结果清晰地揭示了人类CBL在B细胞免疫中的核心且非冗余的功能。
患者临床表现与T细胞功能
在对11名CBL-LOH患者的长期随访中,研究团队发现了一个显著的表型:高达73%(8/11)的患者经历了异常严重的感染,其中两例(P10和P11)为致命性感染。令人惊讶的是,尽管部分患者T细胞胸腺输出略有下降,但成熟T细胞亚群的数量和功能几乎完全正常。患者的CD4+和CD8+ T细胞在体外刺激下能正常增殖和存活,T细胞株在响应IL-2、IL-27刺激时,STAT5磷酸化以及IFN-γ、TNF的产生均正常。这些结果表明,与小鼠模型不同,人类T细胞的发育和功能对CBL的缺失具有强大的代偿能力。
CBL缺失导致B细胞发育异常与功能障碍
与T细胞形成鲜明对比,CBL缺陷患者的B细胞谱系出现了严重紊乱。患者外周血B细胞总数呈现年龄依赖性特征:15岁以下儿童高于或处于正常上限,而15岁以上则降至正常范围以下,提示存在渐进性B细胞减少症。所有患者均表现为多克隆性高丙种球蛋白血症(图3b)。
质谱流式和流式细胞术分析进一步揭示了B细胞亚群的具体变化。在儿童患者中,过渡性B细胞(Transitional B cells)的数量惊人地增加了10.4倍,这些细胞大量富集在未成熟的T1阶段(CD38高表达,CD21低表达),占过渡性B细胞的比例超过50%,而在健康供体中这一比例低于15%(图3c-f)。同时,初始B细胞和记忆B细胞的比例则显著减少(图3d)。对患者骨髓的分析也证实了这种发育阻滞,显示未成熟B细胞(CD34-CD19+CD10+CD20+)比例增加,而前B细胞(pre-BI)比例减少。
通过体外HSPC分化模型,研究者证实了这一发育缺陷是B细胞自主性的。CBL编辑的HSPCs在分化过程中同样出现了未成熟B细胞的积累(图4c,d)。进一步研究发现,CBL缺陷的B细胞对CD38介导的凋亡信号不敏感(图4h,i),并表现出ERK和AKT信号通路的异常激活。此外,从患者体内直接分离出的B细胞在体外分泌免疫球蛋白的能力严重受损,尤其是对T细胞非依赖性刺激的反应更为明显。通过等位基因频率分析发现,在患者体内,携带野生型CBL等位基因的记忆B细胞比例远高于过渡性B细胞,这强有力地证明了CBL缺陷会内在性地阻碍B细胞向记忆细胞分化。
CBL缺陷破坏体液免疫记忆与B细胞耐受
对疫苗接种后抗原特异性B细胞反应的分析显示,尽管CBL缺陷患者能产生SARS-CoV-2刺突蛋白特异性B细胞,但它们的长期维持能力显著受损。在疫苗接种6-12个月后,健康供体的抗原特异性B细胞比例增加了约3倍,相比之下,在疫苗接种后12个月和24个月检测时,同一CBL缺陷个体中检测到的刺突结合B细胞比例与早期时间点相比无明显变化或降低。
同时,这些特异性B细胞发生IgG类别转换的比例也显著降低。BCR库分析进一步揭示了分子层面的缺陷,CBL缺陷患者的记忆B细胞中,体细胞高频突变频率显著降低,且抗体基因的替换/沉默比值降低,表明抗体亲和力成熟过程受损。
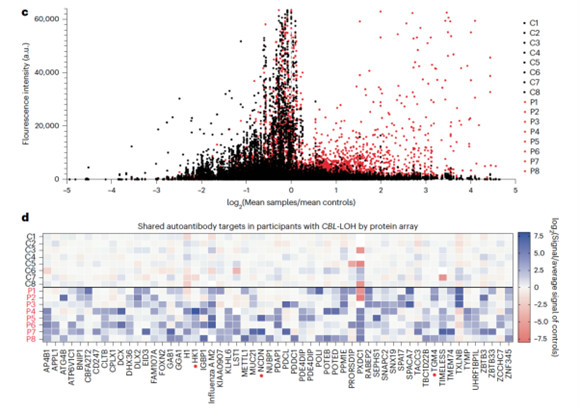
最令人瞩目的是,CBL缺陷患者出现了广泛的自身免疫表型。利用9G4单克隆抗体(识别自身反应性IGHV4-34编码的抗体)染色发现,CBL缺陷患者高达65%的过渡性B细胞、约50%的初始B细胞被9G4抗体“染色”,而在健康供体中这一比例极低,这提示患者血浆中可能存在大量由IGHV4-34基因编码的自身抗体,并覆盖在非自身反应性B细胞表面。使用HuProt人类蛋白质组芯片和InnoScan扫描仪进行的系统性自身抗体筛选证实了这一点,所有接受测试的患者血浆中,针对多种人类自身抗原(如TXLNb等)的IgG抗体反应性均显著增强(图7c,d)。这表明CBL缺陷导致B细胞中枢耐受检查点严重失效,大量自身反应性B细胞得以逃逸并分化成抗体分泌细胞。
讨论和展望
本研究通过对人类CBL缺陷患者的深入剖析,揭示了人与小鼠在免疫调控机制上存在深刻的物种差异。在人类中,CBL并非冗余的T细胞调节因子,而是B细胞发育、耐受和功能所必需的、不可替代的关键分子。CBL的缺失会导致B细胞在过渡期发育阻滞、对CD38介导的凋亡抵抗、B细胞受体信号通路(尤其是PI3K通路)的异常激活,最终导致体液免疫缺陷和自身免疫病的共存。
这一发现具有重要的临床和科学意义。首先,它为解释部分原发性免疫缺陷病患者(尤其是那些表现为B细胞减少、抗体产生障碍,但同时伴有自身免疫症状的患者)的病因提供了新思路。其次,研究指出CBL缺陷与PIK3CD功能获得性突变所导致的活化PI3Kδ综合征(APDS)在B细胞表型上高度相似,包括未成熟B细胞积累、记忆B细胞减少、自身抗体产生等。鉴于已有PI3K抑制剂(如leniolisib)在APDS治疗中显示出良好疗效,本研究提示这些药物可能同样适用于CBL缺陷患者的治疗,为这类罕见疾病提供了有前景的“老药新用”策略。
展望未来,该研究不仅阐明了CBL在人类B细胞中的关键作用,更凸显了在转化免疫学研究中直接研究人类遗传病的必要性和重要性。下一步的研究需要深入探讨CBL如何与CBL-B等其他分子协同作用,共同精细调控B细胞受体信号转导的时空动态。此外,开发能够特异性恢复CBL缺陷B细胞功能或抑制其异常信号通路的靶向治疗药物,将是未来将这一基础研究成果转化为临床应用的最终目标。该研究有力地提醒我们,小鼠模型虽然重要,但人类免疫系统的独特性和复杂性不容忽视。
在免疫系统的复杂调控网络中,E3泛素连接酶CBL(Casitas B-lineage lymphoma)一直被认为是T细胞发育和功能的关键调节因子。此前基于小鼠模型的研究明确指出,CBL对T细胞受体信号具有重要的负调控作用,其缺失会导致T细胞发育异常和功能亢进。然而,CBL在人类免疫系统中的真实角色,特别是其对B细胞的影响,长期以来都未能得到直接证实,因为小鼠研究显示CBL单独缺失并不影响B细胞功能,其作用被其同源蛋白CBL-B所冗余替代。
2026年1月15日发表于《Nature Immunology》的一项突破性研究,彻底颠覆了这一传统认知。由Stuart G. Tangye和Jonathan Bohlen领导的国际联合研究团队,通过对一群携带体细胞CBL功能缺失突变的罕见患者进行长达数年的深入分析,揭示了CBL在人类免疫系统中一个此前被完全忽略的核心作用。与小鼠模型截然相反,人类CBL对T细胞的功能影响甚微,但对于B细胞的发育、成熟、耐受性建立以及体液免疫记忆的形成而言,CBL却是不可或缺的。该研究不仅首次在人体内证实了CBL在B细胞谱系中的非冗余功能,还为理解某些原发性免疫缺陷病和自身免疫病的发病机制提供了全新的视角,并提出了潜在的治疗新策略。
方法
本研究是一项结合了临床队列分析、多组学技术、体外细胞模型和基因编辑技术的综合性研究。研究团队建立了一个由来自9个家庭的11名具有CBL杂合性缺失(LOH)的个体组成的独特队列,此外还包括8名携带遗传性杂合CBL UbLOF变异的个体(包括家庭成员及无亲缘关系者)。为了深入探究CBL缺失对B细胞发育的细胞自主性影响,研究团队运用了先进的基因编辑技术。他们从健康脐带血中分离出CD34+造血干/祖细胞(HSPCs),并使用CRISPR-Cas9系统对其进行编辑,构建了与患者基因型一致的CBL功能缺失突变模型。通过体外诱导分化体系,将这些编辑后的HSPCs向B细胞定向分化,从而在等基因背景下直接观察CBL缺失对早期B细胞发育的直接影响。此外,研究还通过腺嘌呤碱基编辑技术构建了PIK3CD功能获得性突变细胞作为对照,并通过RNA测序比较了两者在转录组水平上的异同。
在B细胞功能研究方面,研究者使用了多种精细的分析手段。通过质谱流式细胞技术对患者外周血中B细胞亚群进行了高维度的免疫表型分析。为了评估B细胞对特定抗原的反应能力,研究人员利用生物素标记的SARS-CoV-2刺突蛋白与链霉亲和素-荧光染料结合,制备了四聚体,用于检测和计数疫苗接种后外周血中抗原特异性B细胞的数量及其类别转换情况。同时,通过流式细胞术分选出不同发育阶段的B细胞亚群(过渡性B细胞、初始B细胞、记忆B细胞),并在体外给予不同刺激(如CD40L/IL-21、BCR交联、CpG等),通过酶联免疫吸附试验(ELISA)检测其分泌免疫球蛋白的能力。
尤为关键的是,本研究利用生物芯片技术对患者血浆中的自身抗体谱进行了高通量筛选。研究人员采用了HuProt人类蛋白质组芯片,该芯片上固定了超过20,000种人类重组蛋白。将患者血清与芯片孵育后,使用Alexa Fluor 647标记的抗人IgG抗体进行检测,随后通过InnoScan 1100AL荧光扫描仪对芯片进行高分辨率扫描。InnoScan 1100AL凭借其高灵敏度的光电倍增管和精确的扫描机制,能够捕捉到微弱的荧光信号,并将其转化为定量的数据。GenePix Pro软件被用于分析图像,提取每个蛋白点的信号强度,从而实现对患者血浆中针对人类自身抗原的IgG抗体进行系统性、高通量的定性及半定量分析。这一技术平台使得研究者能够全面描绘CBL缺陷患者体内自身免疫反应的广谱特征,为揭示其自身免疫表型提供了关键证据。
InnoScan 1100AL荧光芯片扫描仪
结果
研究结果清晰地揭示了人类CBL在B细胞免疫中的核心且非冗余的功能。
患者临床表现与T细胞功能
在对11名CBL-LOH患者的长期随访中,研究团队发现了一个显著的表型:高达73%(8/11)的患者经历了异常严重的感染,其中两例(P10和P11)为致命性感染。令人惊讶的是,尽管部分患者T细胞胸腺输出略有下降,但成熟T细胞亚群的数量和功能几乎完全正常。患者的CD4+和CD8+ T细胞在体外刺激下能正常增殖和存活,T细胞株在响应IL-2、IL-27刺激时,STAT5磷酸化以及IFN-γ、TNF的产生均正常。这些结果表明,与小鼠模型不同,人类T细胞的发育和功能对CBL的缺失具有强大的代偿能力。
CBL缺失导致B细胞发育异常与功能障碍
与T细胞形成鲜明对比,CBL缺陷患者的B细胞谱系出现了严重紊乱。患者外周血B细胞总数呈现年龄依赖性特征:15岁以下儿童高于或处于正常上限,而15岁以上则降至正常范围以下,提示存在渐进性B细胞减少症。所有患者均表现为多克隆性高丙种球蛋白血症(图3b)。
质谱流式和流式细胞术分析进一步揭示了B细胞亚群的具体变化。在儿童患者中,过渡性B细胞(Transitional B cells)的数量惊人地增加了10.4倍,这些细胞大量富集在未成熟的T1阶段(CD38高表达,CD21低表达),占过渡性B细胞的比例超过50%,而在健康供体中这一比例低于15%(图3c-f)。同时,初始B细胞和记忆B细胞的比例则显著减少(图3d)。对患者骨髓的分析也证实了这种发育阻滞,显示未成熟B细胞(CD34-CD19+CD10+CD20+)比例增加,而前B细胞(pre-BI)比例减少。
通过体外HSPC分化模型,研究者证实了这一发育缺陷是B细胞自主性的。CBL编辑的HSPCs在分化过程中同样出现了未成熟B细胞的积累(图4c,d)。进一步研究发现,CBL缺陷的B细胞对CD38介导的凋亡信号不敏感(图4h,i),并表现出ERK和AKT信号通路的异常激活。此外,从患者体内直接分离出的B细胞在体外分泌免疫球蛋白的能力严重受损,尤其是对T细胞非依赖性刺激的反应更为明显。通过等位基因频率分析发现,在患者体内,携带野生型CBL等位基因的记忆B细胞比例远高于过渡性B细胞,这强有力地证明了CBL缺陷会内在性地阻碍B细胞向记忆细胞分化。
CBL缺陷破坏体液免疫记忆与B细胞耐受
对疫苗接种后抗原特异性B细胞反应的分析显示,尽管CBL缺陷患者能产生SARS-CoV-2刺突蛋白特异性B细胞,但它们的长期维持能力显著受损。在疫苗接种6-12个月后,健康供体的抗原特异性B细胞比例增加了约3倍,相比之下,在疫苗接种后12个月和24个月检测时,同一CBL缺陷个体中检测到的刺突结合B细胞比例与早期时间点相比无明显变化或降低。
同时,这些特异性B细胞发生IgG类别转换的比例也显著降低。BCR库分析进一步揭示了分子层面的缺陷,CBL缺陷患者的记忆B细胞中,体细胞高频突变频率显著降低,且抗体基因的替换/沉默比值降低,表明抗体亲和力成熟过程受损。
最令人瞩目的是,CBL缺陷患者出现了广泛的自身免疫表型。利用9G4单克隆抗体(识别自身反应性IGHV4-34编码的抗体)染色发现,CBL缺陷患者高达65%的过渡性B细胞、约50%的初始B细胞被9G4抗体“染色”,而在健康供体中这一比例极低,这提示患者血浆中可能存在大量由IGHV4-34基因编码的自身抗体,并覆盖在非自身反应性B细胞表面。使用HuProt人类蛋白质组芯片和InnoScan扫描仪进行的系统性自身抗体筛选证实了这一点,所有接受测试的患者血浆中,针对多种人类自身抗原(如TXLNb等)的IgG抗体反应性均显著增强(图7c,d)。这表明CBL缺陷导致B细胞中枢耐受检查点严重失效,大量自身反应性B细胞得以逃逸并分化成抗体分泌细胞。
图7c,d
讨论和展望
本研究通过对人类CBL缺陷患者的深入剖析,揭示了人与小鼠在免疫调控机制上存在深刻的物种差异。在人类中,CBL并非冗余的T细胞调节因子,而是B细胞发育、耐受和功能所必需的、不可替代的关键分子。CBL的缺失会导致B细胞在过渡期发育阻滞、对CD38介导的凋亡抵抗、B细胞受体信号通路(尤其是PI3K通路)的异常激活,最终导致体液免疫缺陷和自身免疫病的共存。
这一发现具有重要的临床和科学意义。首先,它为解释部分原发性免疫缺陷病患者(尤其是那些表现为B细胞减少、抗体产生障碍,但同时伴有自身免疫症状的患者)的病因提供了新思路。其次,研究指出CBL缺陷与PIK3CD功能获得性突变所导致的活化PI3Kδ综合征(APDS)在B细胞表型上高度相似,包括未成熟B细胞积累、记忆B细胞减少、自身抗体产生等。鉴于已有PI3K抑制剂(如leniolisib)在APDS治疗中显示出良好疗效,本研究提示这些药物可能同样适用于CBL缺陷患者的治疗,为这类罕见疾病提供了有前景的“老药新用”策略。
展望未来,该研究不仅阐明了CBL在人类B细胞中的关键作用,更凸显了在转化免疫学研究中直接研究人类遗传病的必要性和重要性。下一步的研究需要深入探讨CBL如何与CBL-B等其他分子协同作用,共同精细调控B细胞受体信号转导的时空动态。此外,开发能够特异性恢复CBL缺陷B细胞功能或抑制其异常信号通路的靶向治疗药物,将是未来将这一基础研究成果转化为临床应用的最终目标。该研究有力地提醒我们,小鼠模型虽然重要,但人类免疫系统的独特性和复杂性不容忽视。
相关文章
更多 >