

文献解读:HSP72-PRDX6调控PINK1去泛素化激活线粒体自噬改善MASLD
2026-05-11 来源:本站 点击次数:115
2026年4月1日,海南大学黄玲/饶勇团队联合中山大学黄志纾教授、澳大利亚皇家墨尔本理工大学叶冀明教授等研究团队,在肝病学领域权威期刊Hepatology在线发表了题为 “HSP72 interacts with PRDX6 to deubiquitinate mitochondrial PINK1, activating mitophagy to treat MASLD” 的研究论文。该研究以 MASLD 小鼠模型、原代肝细胞及临床样本为研究对象,系统揭示了HSP72 通过与 PRDX6 相互作用,介导 PINK1 去泛素化并激活线粒体自噬,进而改善 MASLD的全新机制。研究首次证实 PRDX6 是 HSP72 调控线粒体自噬的关键互作蛋白,并鉴定出 PINK1 的 K318 位点是其泛素化降解的关键位点,明确 HSP72/PRDX6/PINK1 轴是 MASLD 进展的重要调控通路。本研究不仅阐明了 MASLD 中线粒体自噬异常的上游分子机制,更为 MASLD 的靶向干预提供了全新的分子靶点与理论依据。
01 研究方法
1.1 实验动物模型构建与处理
研究选用 C57BL/6J 小鼠,构建全身 HSP72 敲除、肝脏特异性 HSP72 敲除、肝脏特异性 PRDX6 敲除、肝脏特异性 HSP72 过表达小鼠,同时设置野生型对照小鼠。采用高脂高糖(HFC)饮食诱导小鼠 MASLD 模型,正常饮食小鼠作为空白对照,按照固定周期进行饲养与样本收集。
1.2 细胞水平实验设计
分离小鼠原代肝细胞,使用油酸(OA)诱导肝细胞脂毒性损伤模拟 MASLD 细胞模型。通过 siRNA 转染实现 HSP72、PRDX6 的敲降,通过质粒转染实现 HSP72、PRDX6 的过表达,同时构建 PINK1 多位点突变质粒用于位点验证。选用 Mitiv-1、CCCP、MG-132、氯喹、PR-619 等工具药,分别干预线粒体分裂、线粒体自噬、蛋白酶体、溶酶体及去泛素化通路。
1.3 分子生物学与形态学检测
通过免疫共沉淀联合液相色谱 - 质谱(Co-IP+LC-MS)筛选 HSP72 的互作蛋白;运用分子对接、分子动力学模拟及结合自由能计算,分析 HSP72 与 PRDX6 的互作模式与稳定性。采用透射电子显微镜(TEM)观察肝脏及肝细胞线粒体形态、线粒体自噬小体与自噬溶酶体形成情况;通过免疫荧光染色检测目标蛋白的表达、定位及共定位情况。利用 Western blot、qPCR 检测线粒体自噬、脂生成、炎症、凋亡相关基因与蛋白的表达水平;检测线粒体 DNA 拷贝数、线粒体耗氧率、线粒体活性氧(mtROS)、丙二醛(MDA)等指标,评估线粒体功能与氧化应激水平。
1.4 临床样本收集与验证
纳入健康人群与 MASLD 患者的肝脏临床样本,排除酒精性肝病、病毒性肝炎等其他肝脏疾病干扰。对临床样本进行病理检测、蛋白表达检测及相关性分析,验证动物与细胞实验的结论。
1.5 统计学分析
采用合适的统计学方法进行数据分析,两组间比较采用 t 检验,多组间比较采用单因素方差分析结合事后检验,非正态分布数据采用非参数检验,明确数据的统计学差异。
02 研究内容
2.1 MASLD 中肝脏线粒体自噬受抑且 HSP72 表达降低
在 MASLD 模型小鼠与 MASLD 患者的肝脏组织中,线粒体自噬水平显著下降,损伤线粒体大量堆积,线粒体自噬小体与自噬溶酶体生成减少,线粒体自噬关键通路 PINK1/Parkin 相关蛋白表达均降低。同时,MASLD 肝脏中 HSP72 的表达水平显著下调,HSP72 表达量与损伤线粒体数量呈负相关,与线粒体自噬水平、线粒体 PINK1 含量呈正相关,提示 HSP72 可能是调控 MASLD 肝脏线粒体自噬的重要分子。
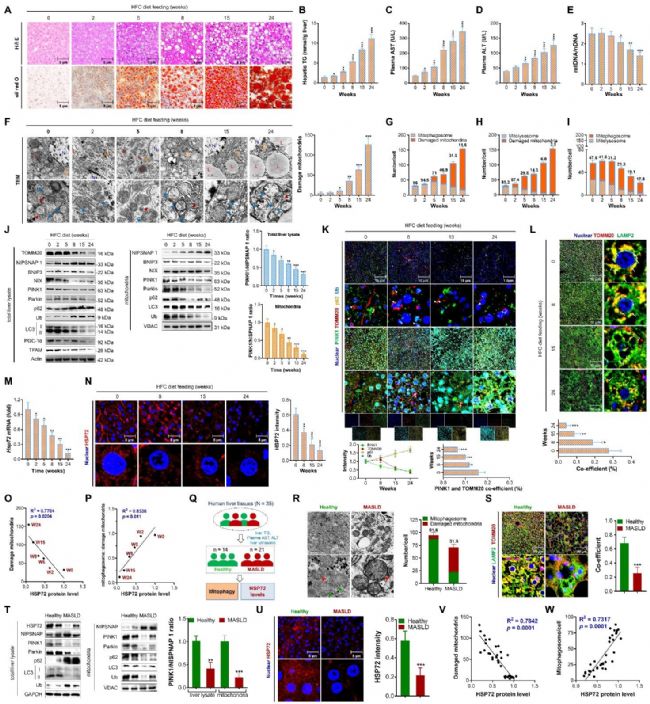
图1 MASLD 小鼠与患者肝脏线粒体自噬水平下降且 HSP72 表达降低
2.2 抑制线粒体自噬可加重小鼠 MASLD 病变
使用线粒体自噬抑制剂处理 MASLD 模型小鼠后,小鼠肝脏线粒体自噬进一步受抑,损伤线粒体堆积加剧,肝脏脂生成相关基因表达上调。同时,小鼠肝脏重量、肝脏甘油三酯含量升高,肝损伤指标水平上升,MASLD 相关病理损伤显著加重,证实线粒体自噬缺陷是 MASLD 进展的重要驱动因素。
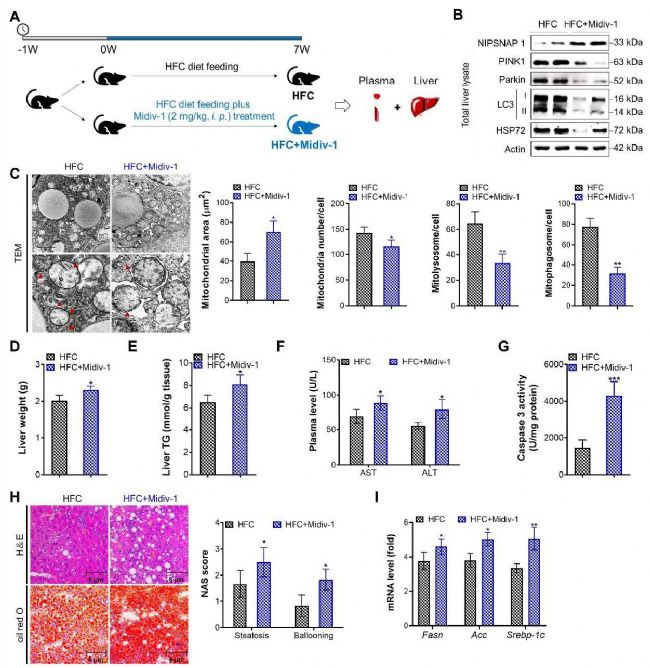
图S5 药物抑制线粒体自噬会进一步加重 MASLD。
2.3 HSP72 缺失抑制线粒体自噬并加重 MASLD
全身敲除 HSP72 的小鼠,在正常饮食与 HFC 饮食诱导下,均出现肝脏脂质堆积、肝损伤加重的表现,脂生成与肝细胞死亡相关基因表达上调。肝脏特异性敲除 HSP72 可得到与全身敲除一致的结果,进一步证实肝脏 HSP72 缺失会导致小鼠肝脏线粒体拷贝数减少、线粒体形态损伤加重,线粒体自噬小体与自噬溶酶体生成受阻,PINK1/Parkin 通路受抑,最终加剧 MASLD 病变。
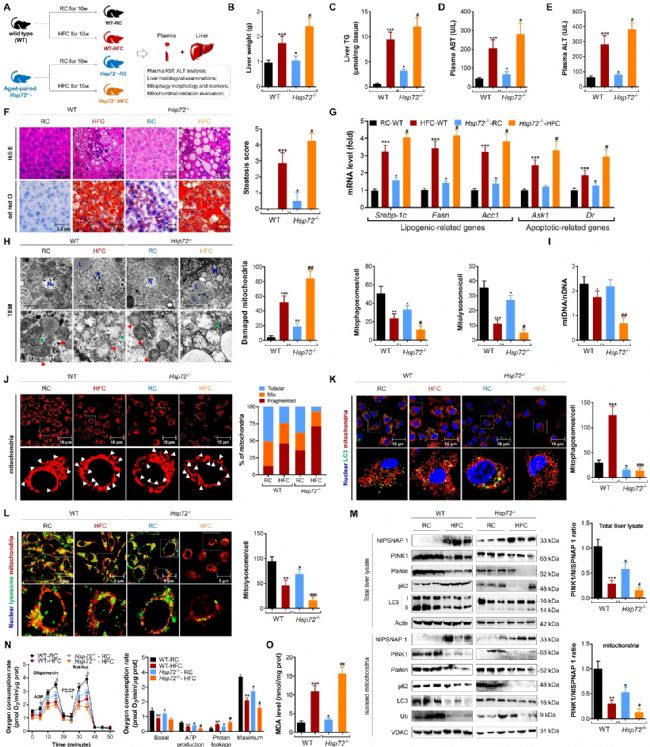
图2 敲除 HSP72 损伤肝脏线粒体自噬并加重小鼠 MASLD
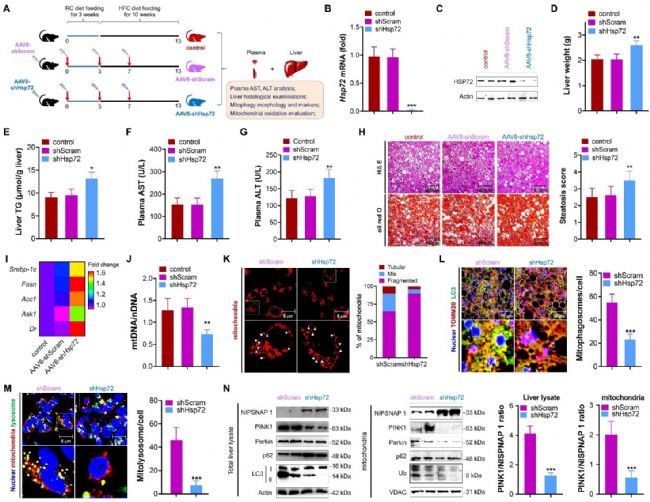
图3 肝脏特异性敲除 HSP72 抑制线粒体自噬并加重小鼠 MASLD
2.4 肝脏过表达 HSP72 激活线粒体自噬并改善 MASLD
在 MASLD 模型小鼠肝脏中过表达 HSP72 后,小鼠肝脏重量、脂质含量、肝损伤指标均显著降低,肝脏病理损伤得到明显缓解。同时,HSP72 过表达可恢复肝脏线粒体拷贝数,改善线粒体形态,促进线粒体自噬小体与自噬溶酶体生成,激活 PINK1/Parkin 介导的线粒体自噬,提升线粒体氧化功能,降低氧化应激水平。在原代肝细胞中过表达 HSP72,也可逆转油酸诱导的线粒体自噬抑制与脂毒性损伤。
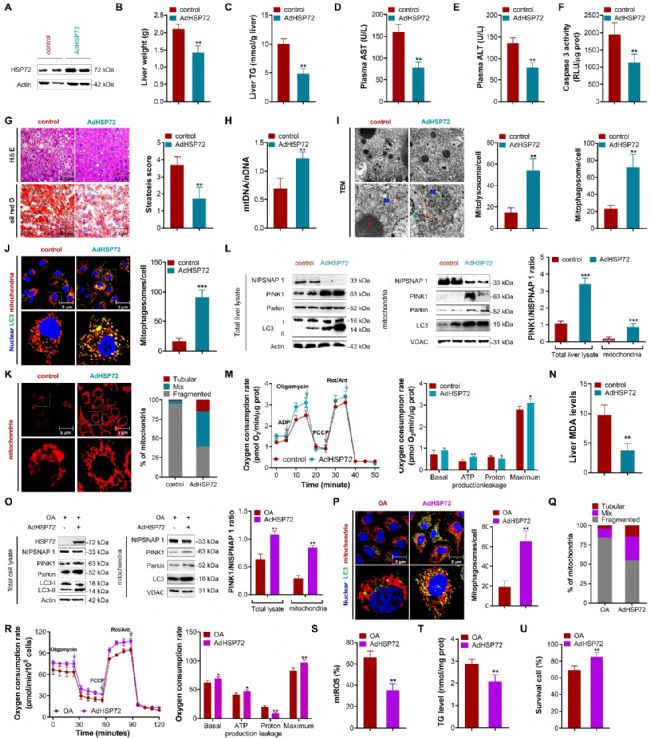
图4 肝脏过表达 HSP72 恢复线粒体自噬并改善小鼠 MASLD
2.5 PRDX6 是 HSP72 调控线粒体自噬的关键互作蛋白
通过免疫共沉淀联合质谱筛选,鉴定出 PRDX6 是 HSP72 的关键互作蛋白,且该蛋白与线粒体自噬调控密切相关。在 MASLD 模型小鼠、MASH 模型小鼠及 MASLD 患者肝脏中,HSP72 与 PRDX6 的相互作用显著减弱,两种蛋白的表达水平均降低。分子模拟结果显示,HSP72 与 PRDX6 可通过氢键、静电作用、疏水作用形成稳定复合物,HSP72 可促进变性 PRDX6 的复性,维持 PRDX6 的稳定性与功能。
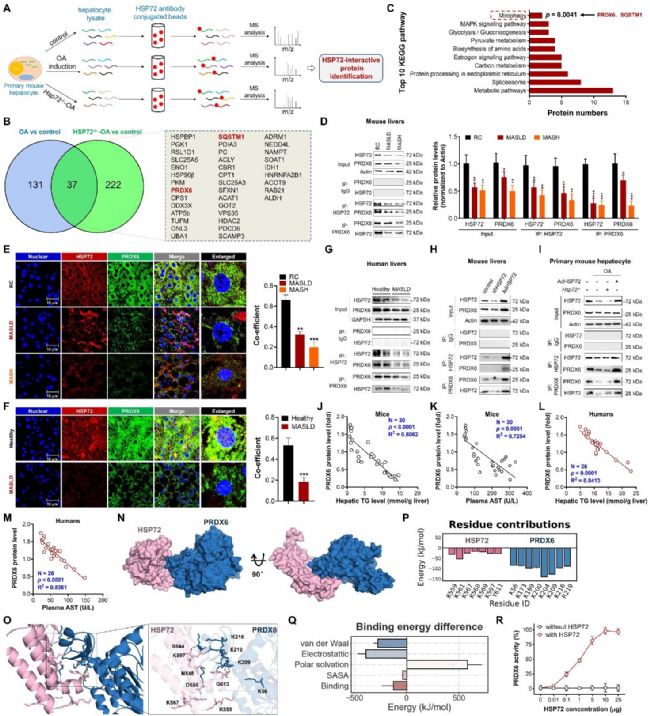
图5 PRDX6 是 HSP72 的互作蛋白并参与线粒体自噬调控
2.6 PRDX6 是线粒体 PINK1 稳定的必需分子
在原代肝细胞中敲除 PRDX6,会导致线粒体形态损伤加剧,线粒体自噬受抑,线粒体 PINK1 含量降低,PINK1/Parkin 通路激活受阻,同时线粒体氧化功能下降、活性氧堆积,脂质堆积与肝细胞损伤加重。而过表达 PRDX6 可逆转上述表型,恢复线粒体 PINK1 水平,激活线粒体自噬,改善肝细胞脂毒性损伤,且 PRDX6 表达量与线粒体 PINK1 水平呈显著正相关。
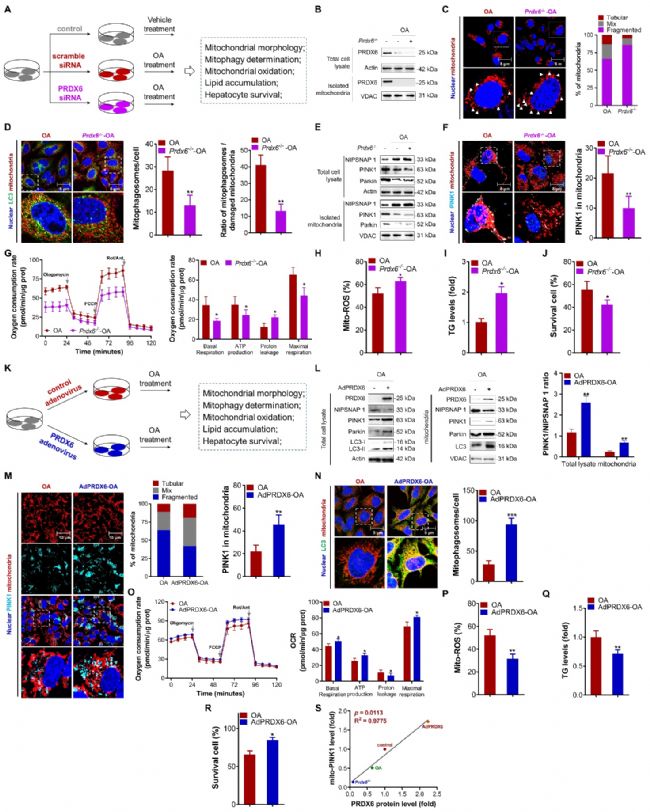
图6 PRDX6 是 PINK1 募集到损伤线粒体的必需因子
2.7 肝脏特异性 PRDX6 缺失阻断线粒体自噬并促进 MASLD
肝脏特异性敲除 PRDX6 后,HFC 饮食诱导的小鼠肝脏线粒体自噬显著受抑,损伤线粒体大量堆积,线粒体氧化功能下降。同时,小鼠肝脏脂质堆积、肝损伤、炎症反应与肝细胞凋亡均显著加重,脂生成、炎症、凋亡相关基因表达全面上调,证实肝脏 PRDX6 缺失会通过抑制线粒体自噬加速 MASLD 进展。
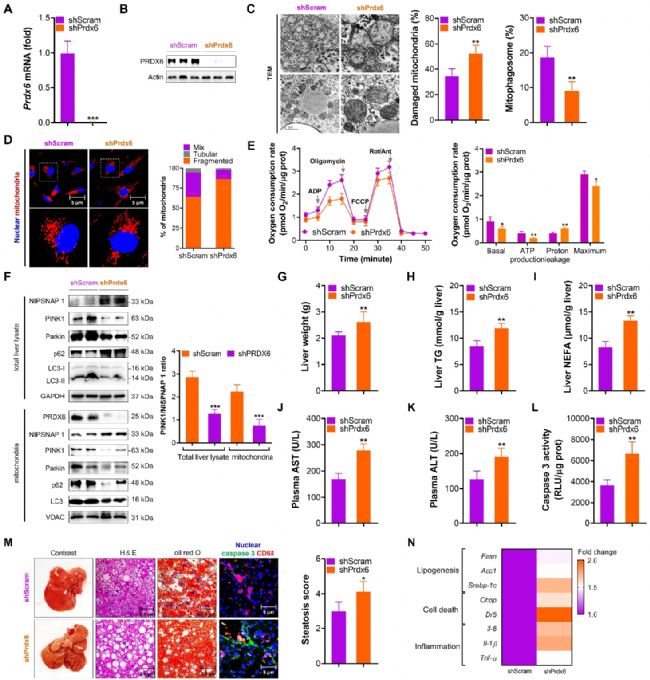
图7 肝脏特异性 PRDX6 缺失抑制线粒体自噬并促进 MASLD
2.8 PRDX6 通过抑制 PINK1 K318 位点泛素化稳定 PINK1
机制研究发现,PRDX6 可降低 PINK1 的泛素化水平,PRDX6 缺失会显著促进 PINK1 泛素化降解。通过位点筛选与突变验证,确定 PINK1 的 K318 位点是 PRDX6 调控的关键泛素化位点,该位点突变可阻断 PINK1 的泛素化,恢复线粒体自噬,改善线粒体功能与肝细胞脂毒性损伤,且该过程依赖去泛素化酶的参与。
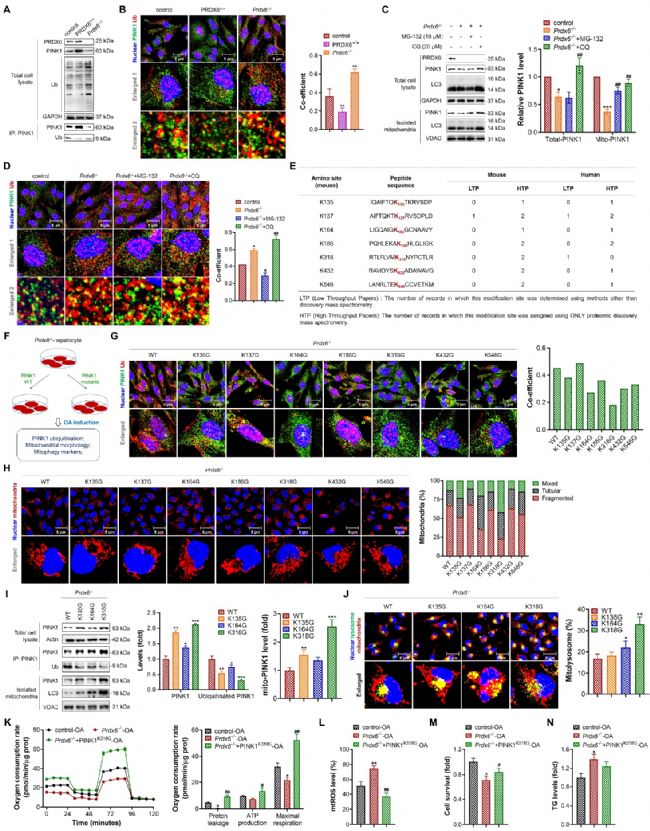
图8 PINK1 的 K318 位点是其泛素化的关键位点,受 PRDX6 调控
2.9 HSP72/PRDX6 轴通过 PINK1 去泛素化激活线粒体自噬改善 MASLD
HSP72 可与 PRDX6 直接结合,促进 PRDX6 的线粒体转位与稳定性;PRDX6 进而靶向 PINK1 的 K318 位点,抑制该位点泛素化,维持线粒体 PINK1 的稳定表达,激活 PINK1/Parkin 依赖型线粒体自噬,清除损伤线粒体,恢复线粒体能量代谢与氧化功能,最终缓解 MASLD 的脂质堆积、炎症与肝损伤。
03 创新点
首次发现并证实HSP72/PRDX6轴是调控 MASLD 肝脏线粒体自噬的全新关键通路,填补了 HSP72 调控肝脏线粒体自噬的分子机制空白。首次鉴定出PINK1 的 K318 位点是其泛素化降解的核心位点,区别于已报道的 K137 位点,完善了 PINK1 翻译后修饰调控线粒体自噬的位点理论。阐明了 HSP72 作为分子伴侣稳定 PRDX6,PRDX6 通过去泛素化维持 PINK1 稳定,进而激活线粒体自噬对抗 MASLD 的完整分子机制,为 MASLD 发病机制提供了新的理论解释。明确 HSP72、PRDX6 及 PINK1 K318 位点可作为 MASLD 治疗的潜在靶点,为 MASLD 的靶向药物开发与基因治疗提供了全新方向。
04 启发
在研究思路上,本研究遵循“临床现象发现 — 动物模型验证 — 细胞机制解析 — 分子互作挖掘 — 关键位点定位 — 临床样本佐证” 的完整逻辑链,是代谢性肝病机制研究的经典范式,为同类代谢性疾病的机制探索提供了可借鉴的思路。在机制拓展上,线粒体自噬是 MASLD 干预的核心靶点,分子伴侣、抗氧化蛋白、激酶的级联调控模式,为线粒体自噬的上游调控研究提供了新的探索方向。在转化应用上,靶向 HSP72/PRDX6/PINK1 K318 通路,可开发小分子激活剂、基因编辑药物等,为 MASLD 的临床治疗提供全新策略。在科研方法上,将多组学筛选、分子模拟、基因编辑、形态学观察、临床样本验证相结合,多维度验证结论,大幅提升了研究的可靠性与创新性,值得在科研工作中学习与应用。
参考文献:
Rao, Yong1; Su, Rui1; Cao, Wen-jie1; Chen, Yue1; Huang, Shu-heng1; Wu, Jun-jie1; Henstridge, Darren C.2; Huang, Lin-sheng3; Liu, Jin1; Liu, Fei-fei1; Jiang, Zhong-ping1; Xu, Cong-jun1; Huang, Zhi-shu4; Ye, Ji-ming5; Huang, Ling1. HSP72 interacts with PRDX6 to deubiquitinate mitochondrial PINK1, activating mitophagy to treat MASLD. Hepatology ():10.1097/HEP.0000000000001763, April 1, 2026. | DOI: 10.1097/HEP.0000000000001763
团队介绍:
面向国家重大需求,海洋、南药(黎药)创新药物研发团队以海南省热带特色药用作物与海洋资源为依托,聚焦“神经退行性疾病与慢性肝病创新药物”两个重大科学难题,结合天然药物化学、合成药物化学、化学生物学、计算机辅助药物设计、药理学、细胞分子生物学等理论及实验技术,在“高活性药物分子指导新靶点发现”与“新靶点指导创新药物设计优化”双重模式驱动下,致力于神经退行性疾病、慢性脂肪肝病等重大疾病的药化药理一体化研究,从而实现新药创制。当前,团队已配备天然药物化学、合成药物化学、药理学与细胞分子生物学等方向领域师资力量。
负责人介绍:
黄玲教授,博士生导师,药学院副院长(挂职)。中山大学药物化学博士,2015年荣获广东省“特支计划”科技创新青年拔尖人才和广州市珠江科技新星专项人才称号,2016年赴美国Scripps研究所神经生物学系访学,2021年通过高层次人才引进到海南大学工作。
针对“高效设计和发现神经退行性疾病”重大科学难题,结合药物化学、化学生物学、神经药理学等理论及实验技术,致力于神经退行性疾病创新药物设计与发现研究,在J. Med. Chem., Eur. J. Med. Chem.等国际期刊发表SCI论文61篇,其中第一作者或通讯作者论文39篇(中科院大类一区Top期刊论文11篇/JCR分区Q1论文19篇),他引2000余次,3篇论文他引超过100次,单篇最高他引151次,h-index 30。申请中国发明专利15项,已获授权专利4项,其中三项专利成果已与企业签订新药合作开发协议。
研究方向:
(1)海洋与南药(黎药)来源的先导化合物发现;
(2)小分子化合物设计、合成与优化;
(3)抗神经退行性疾病与慢性脂肪肝病新靶点与新机制研究。
团队科研成果:
肝病药物研究
发现海洋真菌来源的抗肝纤维化先导化合物(Chrysogenolide D),IC₅0 达 0.7μM。
揭示L - 天冬氨酸通过代谢轴治疗急性肝损伤的新机制。
开发海洋产物HN-001,有效治疗小鼠非酒精性脂肪肝。
神经退行性疾病药物研究
从南海真菌中发现抗阿尔茨海默病的聚酮与倍半萜类化合物,靶向抑制神经炎症。
设计并合成高选择性铁死亡诱导剂(靶向 GPX4),用于恶性肿瘤治疗。
厚谱实验室(脑体互作研究—神经药效评价实验室)专注于脑体互作机制解析与神经药效精准评价的尖端实验技术高地。可以开展清醒动物小分子取样分析、神经递质代谢产物实时分析、癫痫睡眠脑电采集分析、在体电生理记录分析、脑机接口方案评价(大动物脑部手术)、神经调控、组织透明化、脊髓损伤康复验证实验电生理(SEP)、脑立体定位给药、自动无接触采血分析、葡萄糖钳夹实验、行为学迷宫实验和动物造模等各种动物实验服务!
-END-


想了解更多内容,获取相关咨询请联系
电 话:+86-0731-84428665
伍经理:+86-180 7516 6076
工程师:+86-180 7311 8029
邮 箱:consentcs@163.com
