

小鼠模型在揭示脂肪肝加重高血糖的"肝-肠轴"独立通路中的应用
2026-07-10 来源:本站 点击次数:212026年6月17日,四川大学华西医院代谢与衰老研究室陈海洋教授团队联合胃肠外科陈亿教授团队在Cell Metabolism(IF: 37)在线发表题为"Hepatocyte-to-intestinal stem cell remote communication regulates blood glucose homeostasis"的研究论文。该研究揭示了一条此前未被充分认识的"肝-肠"远程通讯轴:脂肪肝中过量分泌的碱性磷酸酶(alkaline phosphatase, ALP)经血液循环作用于肠道干细胞(intestinal stem cells, ISCs),通过α2δ-1/Cav1.2-钙调磷酸酶-NFATC2信号轴抑制SOX21表达,阻碍ISCs向L细胞分化,导致肠源降糖激素GLP-1和PYY减少,进而加重高血糖。

南模生物为该研究提供了ALB-CreERT2(目录号:NM-KI-00002)小鼠模型。
肝脏是血糖稳态调控的核心器官,通过糖原储存、糖异生和糖原分解维持血糖平衡。代谢功能障碍相关脂肪性肝病(metabolic dysfunction-associated steatotic liver disease, MASLD),即通常所说的脂肪肝,是全球最常见的代谢性肝病,影响近三分之一的成年人口。已有流行病学研究证实,脂肪肝患者发生2型糖尿病(type 2 diabetes mellitus, T2DM)的风险是正常人群的两倍以上。
传统观点认为,脂肪肝中肝细胞胰岛素抵抗导致肝脏糖异生异常增强,是脂肪肝增加糖尿病风险的主要机制。然而,临床实践中的一个现象提示了更复杂的调控机制:二甲双胍(metformin)作为肝脏糖异生的药物抑制剂,在部分伴有脂肪肝的T2DM患者中并不能有效缓解高血糖,而这些患者在加用GLP-1受体激动剂后血糖得到进一步改善。GLP-1是由肠道L细胞分泌的一种肠源降糖激素,而L细胞的持续补充高度依赖ISC的分化。肝脏是否通过影响ISC的分化命运来调控血糖稳态,此前尚不清楚。
1 脂肪肝对肠道L细胞和血糖稳态的影响
研究团队首先纳入一项包含238例减重手术患者的前瞻性队列(图1a),通过肝活检评估肝脏脂肪变性,并匹配术前空腹血浆样本。结果显示,脂肪肝患者血浆GLP-1水平显著低于正常肝脏人群(图1b)。即便在脂肪肝患者内部,合并T2DM者的血浆GLP-1水平也显著低于不合并T2DM者(图1c)。
为进一步验证,研究团队采用高脂饮食(high-fat diet, HFD)和胆碱缺乏氨基酸限定(choline-deficient L-amino acid-defined, CDAA)饮食分别诱导两种脂肪肝小鼠模型。CDAA饮食模型实验流程如图1d所示,油红O和H&E染色确认了肝脏脂肪变性的成功建立(图1e)。与对照组相比,CDAA诱导的脂肪肝小鼠体重无显著差异,但空腹血糖显著升高且糖耐量受损(图1f-h),同时血浆GLP-1和PYY水平显著降低(图1i, j)。值得注意的是,二甲双胍虽可降低脂肪肝小鼠的血糖并改善糖耐量,但对血浆GLP-1和PYY水平无显著影响(图1f-j)。
为探究脂肪肝影响肠道L细胞的细胞学机制,研究团队对正常饮食和CDAA诱导脂肪肝小鼠的结肠组织进行了单细胞转录组测序(图1k)。整合分析鉴定出五大类结肠上皮细胞类型,发现脂肪肝小鼠肠内分泌细胞(enteroendocrine cells, EECs)比例显著下降(图1l)。进一步对EECs亚群进行分析,发现L细胞比例的减少最为突出(图1m, n)。
研究团队还在一项纳入29例右半结肠癌患者的回顾性队列中进行了验证(图1o),通过腹部CT评估脂肪肝状况。免疫染色结果显示,脂肪肝患者正常近端结肠组织中GLP-1+和PYY+ L细胞数量均显著低于正常肝脏患者(图1p, q)。
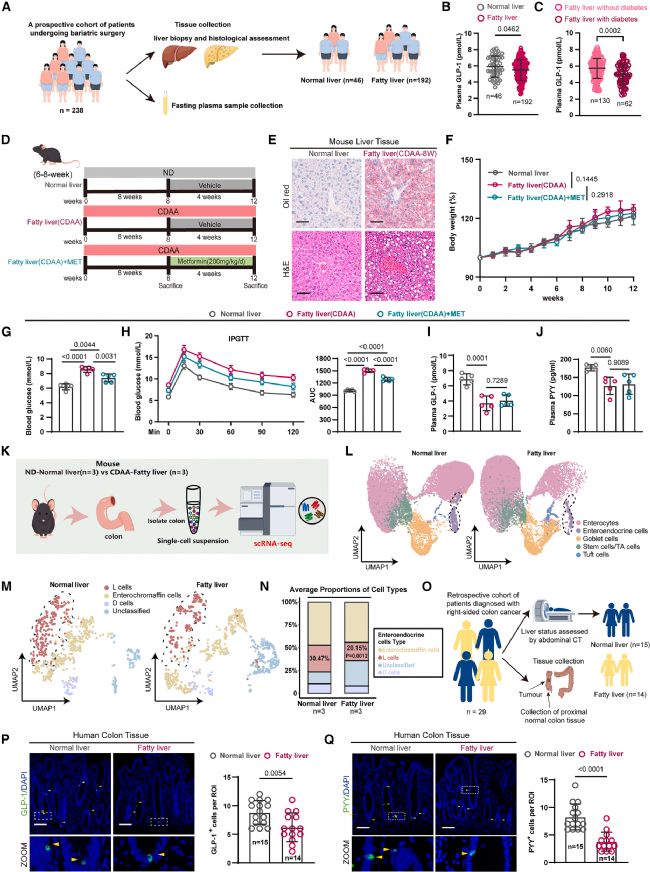
图1. 脂肪肝对肠道L细胞和血糖稳态的影响
2 脂肪肝抑制肠道干细胞向L细胞的分化
研究团队进一步确认了两种脂肪肝小鼠模型中L细胞数量的减少。免疫荧光染色显示,HFD诱导的脂肪肝小鼠结肠GLP-1+(图2a)和PYY+(图2b)L细胞数量均显著减少;CDAA诱导的模型中观察到了一致的表型(图2c, d)。体外培养的结肠类器官也表现出L细胞减少(图2e, f)。在上述所有实验中,二甲双胍治疗均无显著影响(图2a-f)。
为确定L细胞减少的原因,研究团队首先排除了凋亡因素:TUNEL染色显示脂肪肝小鼠结肠上皮凋亡细胞数量与对照组无显著差异(图2g)。基于单细胞转录组数据的拟时序分析(pseudotime analysis)显示,正常肝脏小鼠中ISC向L细胞的分化轨迹清晰可辨(图2h),而脂肪肝小鼠的这一分化轨迹受到抑制(图2i)。
研究团队进一步利用谱系追踪(lineage tracing)技术在体内动态追踪ISC的分化过程(图2j)。结果显示,脂肪肝小鼠中Lgr5+谱系追踪克隆分化为GLP-1+(图2k)和PYY+(图2l)L细胞的比例均显著低于对照组。二甲双胍对ISC向L细胞的分化无显著影响(图2k, l)。
综上,这些结果表明脂肪肝通过抑制ISC向L细胞分化,减少肠道L细胞数量,进而降低血浆GLP-1和PYY水平,损害血糖稳态。
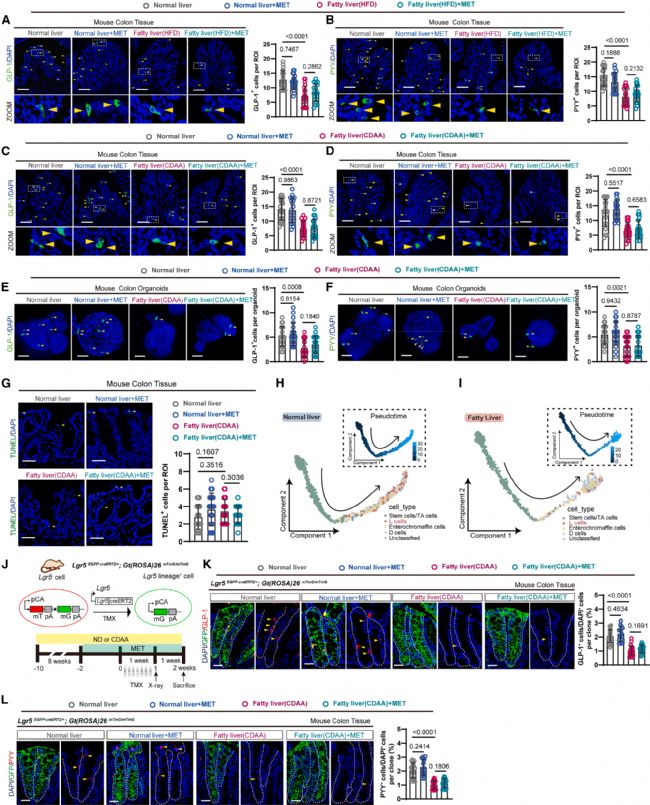
图2. 脂肪肝抑制肠道干细胞向L细胞的分化
3 脂肪肝来源的碱性磷酸酶减少肠道L细胞丰度
脂肪肝患者常伴有肝功能生物标志物的升高,但这些标志物的下游生物学功能尚不明确。研究团队基于前述238例前瞻性队列数据,分析了血浆GLP-1与多项肝功能指标的相关性。结果显示,GLP-1与ALP呈显著负相关(图3a),而与AST、ALT、GGT等指标无显著相关性。
为验证ALP对L细胞的直接效应,研究团队从人结肠隐窝培养肠道类器官,分别添加ALP、AST、ALT或GGT处理(图3b)。结果显示,ALP处理可显著降低类器官中GLP-1+(图3c)和PYY+(图3d)L细胞数量,而AST、ALT和GGT对L细胞丰度无明显影响。
体内实验显示,CDAA诱导的脂肪肝小鼠血浆ALP水平显著升高(图3e)。研究团队通过AAV介导肝脏特异性Alpl(编码ALP蛋白)过表达(图3f),使正常饮食小鼠的血浆ALP水平显著升高(图3g)。与对照组相比,Alpl过表达小鼠结肠中GLP-1+(图3h)和PYY+(图3i)L细胞数量均显著减少。谱系追踪进一步证实,Alpl过表达小鼠中Lgr5+谱系追踪克隆分化为GLP-1+(图3j)和PYY+(图3k)L细胞的比例显著降低。伴随L细胞减少,Alpl过表达小鼠的血浆GLP-1(图3l)和PYY(图3m)水平下降,空腹血糖升高(图3n),糖耐量受损(图3o)。
此外,研究团队在无菌(germ-free)小鼠中进行了肝Alpl过表达,仍观察到类似的L细胞减少和GLP-1/PYY下降表型,表明ALP对ISC分化的调控不依赖于肠道菌群。
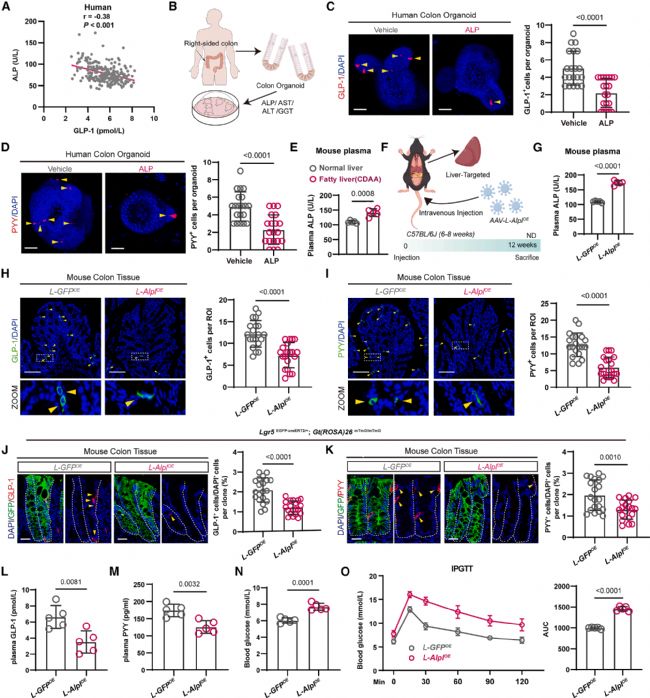
图3. 肝源性ALP减少肠道L细胞数量并损害血糖稳态
4 抑制脂肪肝中ALP表达改善血糖稳态
研究团队通过AAV介导抑制脂肪肝小鼠肝脏Alpl表达。与对照脂肪肝小鼠相比,Alpl抑制后结肠GLP-1+(图4a)和PYY+(图4b)L细胞数量显著增加,血浆GLP-1(图4c)和PYY(图4d)水平回升,空腹血糖降低(图4e),糖耐量改善(图4f)。值得注意的是,在正常肝脏小鼠中抑制Alpl表达对上述指标无显著影响(图4a-f)。
研究团队进一步构建了肝细胞特异性Alpl条件性敲除小鼠(ALB-Cre; Alplfl/fl)。与对照脂肪肝小鼠相比,Alpl敲除的脂肪肝小鼠结肠GLP-1+(图4g)和PYY+(图4h)L细胞显著增多,血浆GLP-1(图4i)和PYY(图4j)升高,血糖降低(图4k),糖耐量改善(图4l)。在正常肝脏小鼠中,Alpl敲除对上述指标同样无显著影响(图4g-l)。
研究团队还检验了抑制肝脏ALP与二甲双胍的协同降糖效应。在HFD诱导的脂肪肝模型中,单独抑制肝脏Alpl或单独使用二甲双胍均可显著降低血糖并改善糖耐量;二者联合使用产生了叠加效应,降糖效果优于任一单独干预(图4m, n)。CDAA诱导的脂肪肝模型中也观察到了一致的结果(图4o, p)。这些结果表明,脂肪肝通过过量分泌ALP损害血糖稳态(图4q),该机制独立于传统的肝脏糖异生增强途径,抑制肝脏ALP表达可独立降糖并与二甲双胍协同增效。
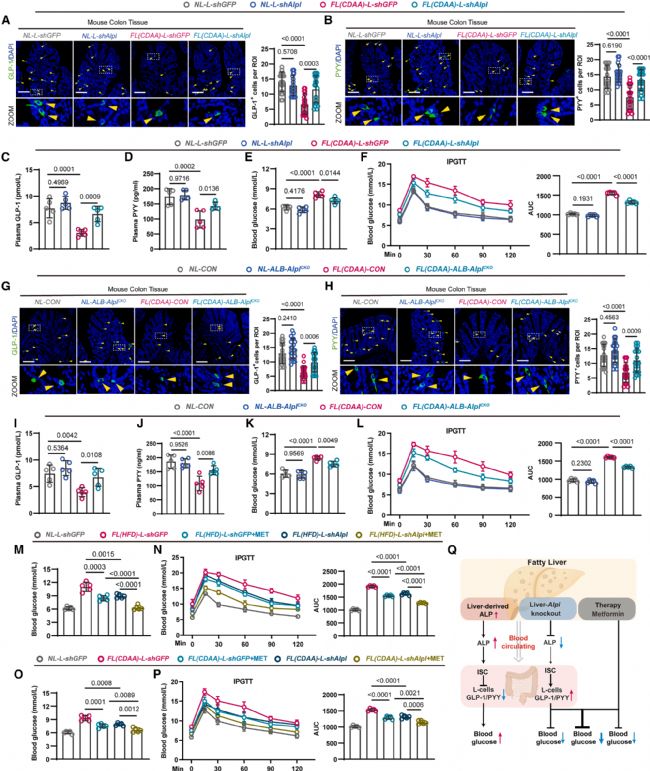
图4. 抑制脂肪肝中ALP表达改善血糖稳态
5 肝源性ALP抑制肠道SOX21诱导高血糖
为探究肝源性ALP影响肠道L细胞丰度的分子机制,研究团队对正常肝脏、脂肪肝和脂肪肝+Alpl抑制三组小鼠的结肠隐窝进行了转录组测序。PCA分析显示,脂肪肝+Alpl抑制组的基因转录谱更接近正常肝脏组,提示Alpl抑制在一定程度上恢复了转录状态(图5a)。GSEA分析发现,Wnt和TGF-β信号通路在脂肪肝+Alpl抑制组中较脂肪肝组显著上调(图5b)。
PPI网络分析将SOX21识别为连接Wnt和TGF-β通路的核心节点(图5c)。基于单细胞转录组数据的UMAP分析确认Sox21主要表达于ISC/TA细胞区域(图5d, e),且脂肪肝小鼠ISCs中Sox21的表达水平显著低于正常肝脏小鼠(图5f)。免疫印迹实验进一步验证,脂肪肝小鼠结肠Lgr5+细胞中SOX21蛋白水平显著降低(图5g),而肝脏Alpl过表达同样导致ISCs中SOX21水平下降(图5h)。
功能层面,研究团队通过AAV在ISC中过表达Sox21。结果显示,在CDAA诱导的脂肪肝小鼠中,Sox21过表达显著增加了结肠GLP-1+(图5i)和PYY+(图5j)L细胞数量,升高了血浆GLP-1(图5k)和PYY(图5l)水平,降低了空腹血糖(图5m)并改善了糖耐量(图5n)。在正常肝脏小鼠中,Sox21过表达虽可增加L细胞数量,但对血糖和糖耐量的影响相对较小(图5i-n)。
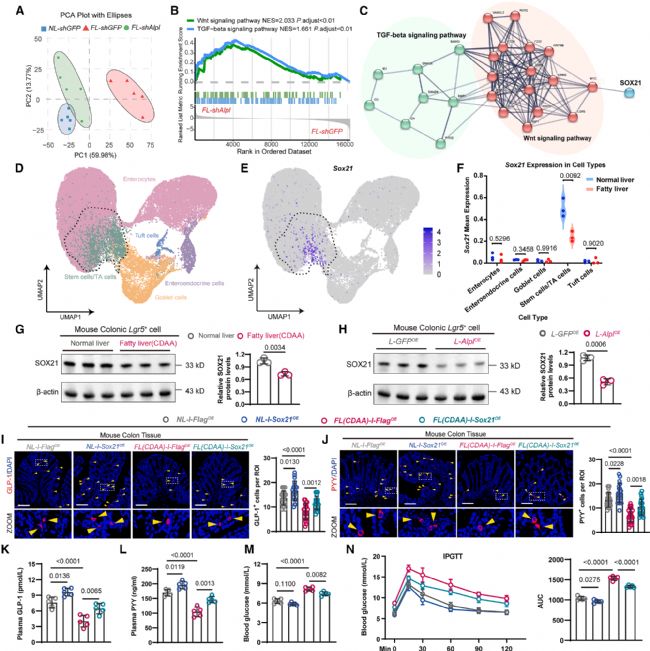
图5. 肝源性ALP抑制肠道SOX21诱导高血糖
6 SOX21是ALP下游调控L细胞分化的关键因子并能转录激活Bmp7
研究团队构建了ISC特异性Sox21条件性敲除小鼠(Lgr5-Sox21CKO)。在正常饮食条件下,与对照小鼠相比,Sox21敲除即导致结肠GLP-1+(图6a)和PYY+(图6b)L细胞数量显著减少,血浆GLP-1(图6c)和PYY(图6d)水平下降,血糖升高(图6e),糖耐量受损(图6f)。
关键的遗传上位性实验进一步验证了ALP与SOX21的调控关系。在CDAA诱导的脂肪肝小鼠中,AAV介导抑制肝脏Alpl表达可显著增加L细胞数量、升高血浆GLP-1和PYY、降低血糖并改善糖耐量;而同时敲除ISC Sox21后,上述改善效应显著减弱(图6g-l)。在脂肪肝小鼠中单独敲除ISC Sox21对上述指标无显著影响(图6g-l)。这些结果确定了ALP通过调控ISC中SOX21表达来影响L细胞分化和血糖稳态。
在SOX21调控L细胞分化的转录机制方面,Lgr5-Sox21CKO小鼠结肠隐窝的GSEA分析显示TGF-β信号通路发生显著变化(图6m)。通过JASPAR和MEME数据库预测SOX21的DNA结合基序(图6n),并与转录组差异表达基因交叉比对,在TGF-β通路中鉴定出骨形态发生蛋白7(bone morphogenetic protein 7, BMP7)为唯一重叠候选基因(图6o)。
功能验证显示,在脂肪肝小鼠ISC中过表达Sox21可显著上调Bmp7 mRNA(图6p)、BMP7蛋白(图6q)及其下游效应分子磷酸化SMAD5(图6r)的表达水平。为进一步确认BMP7是SOX21的功能性下游效应器,研究团队在类器官中进行了回补实验:Sox21过表达可显著增加类器官中GLP-1+(图6s)和PYY+(图6t)L细胞数量,而同时敲低Bmp7可显著削弱这一效应。
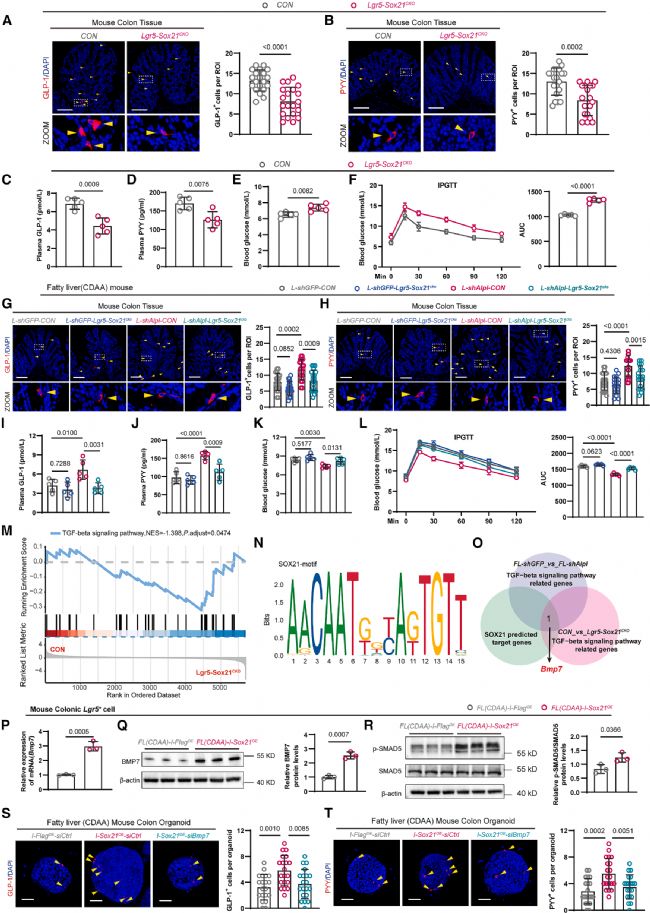
图6. SOX21是ALP下游调控L细胞分化的关键因子并能转录激活Bmp7
7 肝源性ALP结合α2δ-1促进钙内流并通过钙调磷酸酶-NFATC2轴抑制Sox21
在机制链条的最上游,研究团队探究了ALP如何导致ISCs中SOX21表达下降。免疫印迹显示,脂肪肝小鼠结肠Lgr5+细胞膜组分中ALP定位显著增加(图7a),提示ALP可能通过作用于ISC膜来发挥调控功能。研究团队首先排除了ALP磷酸酶活性经典底物(ATP和PPi)对L细胞丰度的影响。
转录组分析提示钙信号通路在脂肪肝小鼠ISCs中差异表达。钙离子浓度测定证实,CDAA诱导的脂肪肝小鼠结肠Lgr5+细胞中细胞内钙离子浓度显著升高,而抑制肝脏Alpl表达可逆转这一升高(图7b)。相反,在正常饮食小鼠中过表达肝脏Alpl则显著增加ISCs中钙离子浓度(图7c)。
通过IP-MS技术分析ISC膜蛋白中ALP的互作蛋白组,电压依赖性钙通道亚基α2δ-1(由Cacna2d1编码)被鉴定为ALP的主要候选结合伙伴,在质谱分析中具有较高的信号强度和独特肽段数(图7d-f)。coIP实验(图7g)和GST pull-down实验(图7h)进一步确认了ALP与α2δ-1之间的直接物理相互作用。
α2δ-1是钙通道Cav1.2(由Cacna1c编码)的关键调节亚基,负责将Cav1.2从胞质转运至细胞膜。在遗传回补实验中,过表达肝脏Alpl可导致ISC膜Cav1.2(图7i)增多和细胞内钙升高(图7j);而在ISC中同时敲低Cacna2d1后,膜Cav1.2增加和钙升高的效应均被消除(图7i, j)。类器官实验中,引入功能缺失突变的α2δ-1后,添加外源ALP无法再增加膜Cav1.2水平(图7k)。相应地,肝脏Alpl过表达导致的GLP-1+(图7l)和PYY⁺(图7m)L细胞减少,在ISC中敲低Cacna2d1后显著恢复。
关于钙信号下游调控SOX21的机制,研究团队通过JASPAR数据库分析Sox21启动子区,预测NFATC2为高亲和力结合转录因子(图7n)。NFATC2是钙-钙调磷酸酶信号通路的关键效应因子。在类器官实验中,ALP处理可减少GLP-1+和PYY+ L细胞数量;而同时使用细胞内钙螯合剂BAPTA-AM或钙调磷酸酶抑制剂环孢素A(CsA)可显著逆转ALP的这一效应,恢复L细胞数量(图7o)。综合上述结果,研究团队提出了一条分子通路模型(图7p):肝源性ALP与ISC膜上α2δ-1结合,促进Cav1.2膜转位和钙内流,激活钙调磷酸酶/NFATC2信号轴转录抑制Sox21,进而阻碍ISC向L细胞分化。

图7. 肝源性ALP结合α2δ-1促进钙内流并通过钙调磷酸酶-NFATC2轴抑制Sox21
此研究揭示了一条不依赖于肝脏糖异生增强的脂肪肝促高血糖新通路:脂肪肝中肝细胞过量分泌ALP进入循环,ALP与ISC膜上α2δ-1结合,促进Cav1.2膜转位和钙内流,激活钙调磷酸酶/NFATC2信号轴转录抑制Sox21,导致BMP7表达下降,阻碍ISC向L细胞分化,最终减少肠源降糖激素GLP-1和PYY的分泌,加重高血糖。研究发现抑制肝脏ALP可独立降低血糖,并与二甲双胍产生协同效应,提示ALP可能作为脂肪肝相关高血糖的潜在干预靶点之一。
研究团队也指出了该工作的若干局限性,包括:尚未解析ALP结合后α2δ-1/Cav1.2复合物的高分辨率三维结构;除ALP外,脂肪肝可能还通过其他分泌因子调控ISC分化;脂肪肝对肠外器官的潜在影响亦有待进一步探究。
相关阅读
疾病模型专辑|糖尿病
STTT | 肝癌细胞为什么疯狂囤积糖原?空军军医大学团队发现AKAP1是背后关键推手
Cell Metabolism | 脂肪肝竟是乳腺癌的“远程指挥官”?科学家揭秘它的致癌信使
全球超1亿患者!MASH药物研发进展与核心模型解析
关于我们
上海南方模式生物科技股份有限公司(Shanghai Model Organisms Center, Inc.,简称"南模生物"),成立于2000年9月,是一家上交所科创板上市高科技生物公司(股票代码:688265),始终以编辑基因、解码生命为己任,专注于模式生物领域,打造了以基因修饰动物模型研发为核心,涵盖多物种模型构建、饲养繁育、表型分析、药物临床前评价等多个技术平台,致力于为全球高校、科研院所、制药企业等客户提供全方位、一体化的基因修饰动物模型产品解决方案。