

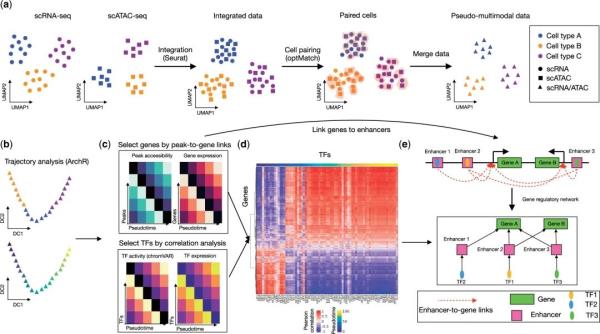
scMega助力scATAC-seq数据与scRNA-seq数据的联合分析
2025-04-09 来源:本站 点击次数:818单细胞RNA测序技术(scRNA-seq)揭示了单个细胞的基因表达情况,单细胞ATAC测序技术(scATAC-seq)专注于单个细胞的染色质开放性,显现了细胞内的基因调控情况。两种技术结合使用可以更好地推断细胞内部的基因调控网络。但是这些数据的分析经常要使用不同分析工具分别进行。例如,对于scRNA-seq数据一般使用Seurat包分析,而scATAC-seq数据则使用ArchR包来进行分析和轨迹推断,对转录因子(transcription factors,TFs)的活性估测则由chromVAR包进行,诸如此类。这使得对单细胞基因调控网络的分析变得十分复杂和不便。
基于这些问题, scMega这样一个整合了多种现有数据分析方法的多组学分析工具应运而生。该工具包含了数据整合、细胞配对、推断伪时间轨迹、TFs筛选、定量基因调控网络和增强子TFs-基因互作识别。
具体而言,scMega可分为三个主要步骤:
①单细胞多组学数据整合,候选TFs和基因的识别与筛选和基因调控网络分析。在单细胞多组学数据整合中,利用Seurat的典型相关分析(CCA)将scRNA-seq数据与scATAC-seq数据进行整合,如果存在批次效应,再利用Harmony进行校正,使用后OptMatch将scRNA-seq与scATAC-seq的细胞进行匹配,构建一个伪多模态数据(图1a)。
②接下来,scMega利用该多模态数据识别候选TFs和基因。首先,使用AchR推断伪时间轨迹(图1b),然后根据染色质可及性谱估计TFs的结合活性,使用chromVAR计算TFs结合活性与TFs表达之间的相关性,具有高相关性说明该TF既高表达,其模体又具有更高的可及性(图1c)。另外,scMega还会根据基因在伪时间轨迹上的表达变化筛选出轨迹相关基因(图1d)。
③最后,在scMega的基因调控网络分析中,当一个基因与至少一个增强子相关联,且某个TF与这些增强子中的至少一个结合时,这个基因被认为是这个TF的靶点,其相互作用按其相关性进行加权,由此得到基于增强子的基因调控网络(图1e)。
而在真实数据的实验中,使用了人类外周血单核细胞的单细胞多模态数据,首先进行数据整合和细胞配对,配对结果虽然真实的细胞对只有少数被配对成功,但同一类型的细胞基本都匹配在一起。随后分别基于真实的细胞对和计算匹配的细胞对进行基因调控网络分析,有75%的TFs,83%的基因和60%的TF-基因调控单元重合,说明大多数真实的互作关系可以由scMega复原。
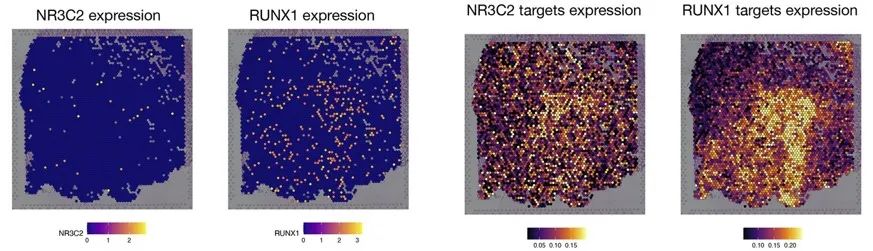
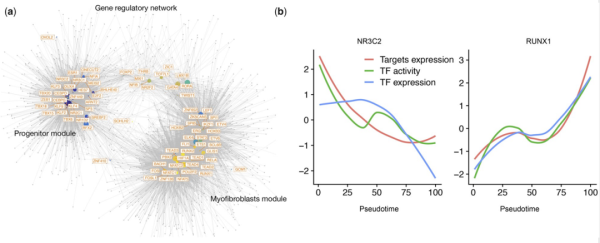
另外的,对人类心脏心肌梗塞后的纤维细胞进行分析,构建了一条在成纤维细胞亚群内的伪时间轨迹,并推断了基因调控网络,识别了祖细胞和肌成纤维细胞亚群内的TF-基因调控对,在空间转录组数据中对这些TF的靶基因进行空间表达检测,也显示了在心脏纤维化区域内靶基因表达存在梯度与互斥的现象(图2)。scMega对于分析结果也具有良好的可视化方法,在网络图中每个节点代表一个TF或靶基因,TF节点的颜色代表了其在伪时间上的位置,而相连接的节点为与该TF相关的靶基因,可见不同的TF在特定细胞亚群中成簇(图3a);在曲线图中,可见不同TF的结合活性、表达和靶基因表达在细胞分化轨迹上的变化(图3b)。
ScMega对于scATAC数据的分析和解读提供了一条非常可行且高效的分析方案,使得scATAC和scRNA数据的联合分析更加简单,如果您上手有scATAC数据而发愁,不妨一试。