

用户文章:DDA PASEF免疫肽组学搜库软件综合评测
2026-06-01 来源:本站 点击次数:288
在精准医疗与肿瘤免疫治疗飞速发展的今天,免疫肽组学(Immunopeptidomics)已成为连接基础生物学与临床转化的关键桥梁。它直接解析被MHC分子呈递到细胞表面的全部抗原肽,是发现肿瘤新抗原、病毒表位、自身免疫抗原、疫苗靶点的最直接、最可靠手段。但免疫肽组学的数据分析,远比常规蛋白质组更具挑战,主要体现在以下几个方面:
软件选择不当会直接导致高置信抗原漏检、假阳性偏高、变异肽识别不准、实验数据无法充分挖掘等问题的发生。
针对这一痛点,德国癌症研究中心免疫肽组学研究平台的Stefan Tenzer教授携团队在 MCP 发表重磅基准测试结果。以JY细胞系MHC Ⅰ/Ⅱ免疫肽为标准样品,基于 Thunder DDA PASEF 采集质谱数据,对当前五大主流搜库软件开展深度对比。
结果表明 PEAKS® 的 DeepNovo Peptidome workflow 对免疫肽组学研究具备强大的综合分析能力,为全球免疫肽组学研究者提供了最直观的软件选择与流程优化依据。
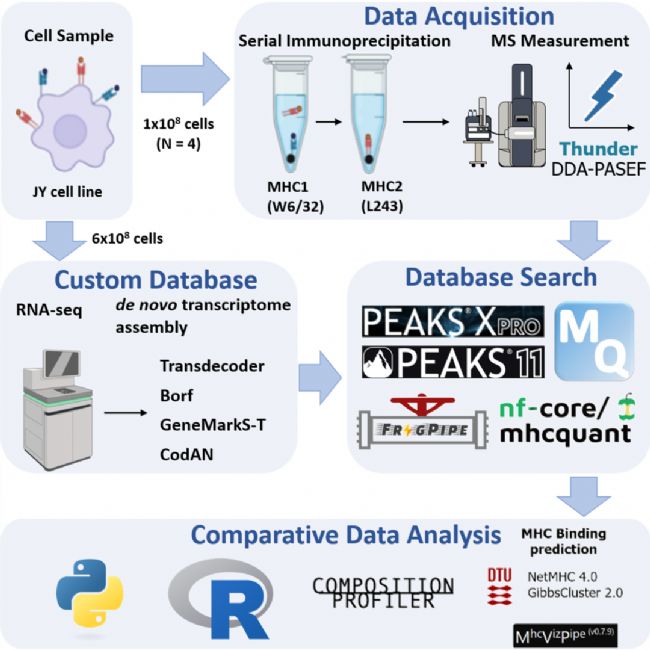
本次基准测试对比了 PEAKS® Online 11, PEAKS Studio Xpro, FragPipe, MaxQuant 和 MHCquant 五个搜库软件,均设置1% PSM FDR。
Figure 1A 的整体统计结果显示,使用PEAKS® Online 11内置的 DeepNovo Peptidome workflow 鉴定到的总数最高,MHC Ⅰ/Ⅱ肽段较 PEAKS Studio X Pro中的传统 DB Search 和 FragPipe 高出 6%–11%。FragPipe 紧随其后在开源软件中表现最佳,MHCquant 表现稳健,略低于 FragPipe,MaxQuant 鉴定量最低,较 MHCquant 少 50%–60%。Figure 1 交集分析显示,除 MaxQuant 外,其他软件间的重合度约 58%-60%,说明主流工具对高置信肽判断一致,整体无极端偏好偏差。Figure 1C-D 表明所有软件对母离子电荷及肽段长度分布均高度一致,无系统性偏差。
(新版 PEAKS ®Online 与 PEAKS® Studio 已实现算法统一,均支持DeepNovo Peptidome的肽组学专属工作流,用户可在任一平台获得同等高性能表现。)
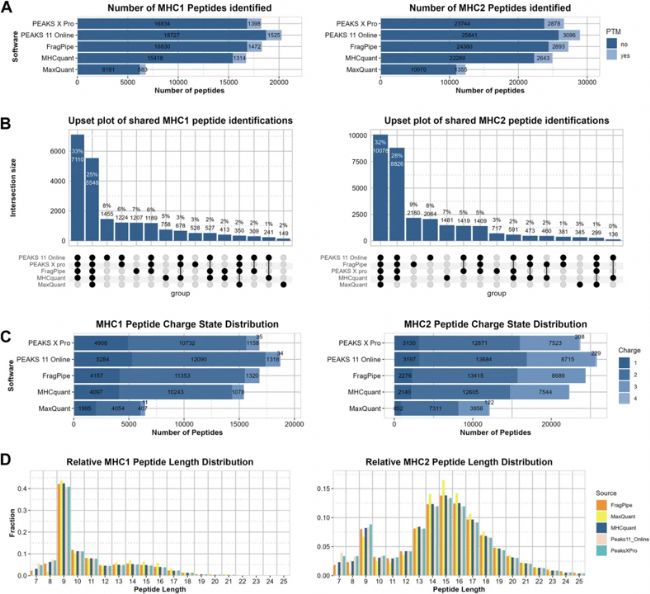
随后,作者对所有软件鉴定到的多肽通过 netMHCpan 和 netMHCIIpan 分别对MHC-Ⅰ和MHC-Ⅱ型多肽进行结合强度预测,Figure 2 结果显示,PEAKS ®DeepNovo Peptidome workflow鉴定到的总结合肽数量最多,覆盖更广;MHCquant 结合肽本身比例更高,更偏向于高特异性结合肽段。各软件氨基酸组成高度保守,且样本重现性基本稳定,仅 MaxQuant 有少量显著偏差。

Figure 2 免疫肽结合力和重现性对比
同时,作者对比了 5 款软件共同鉴定到肽段的置信度得分 Spearman 相关系数,结果显示越多软件共有的多肽,相关性排名越靠前。
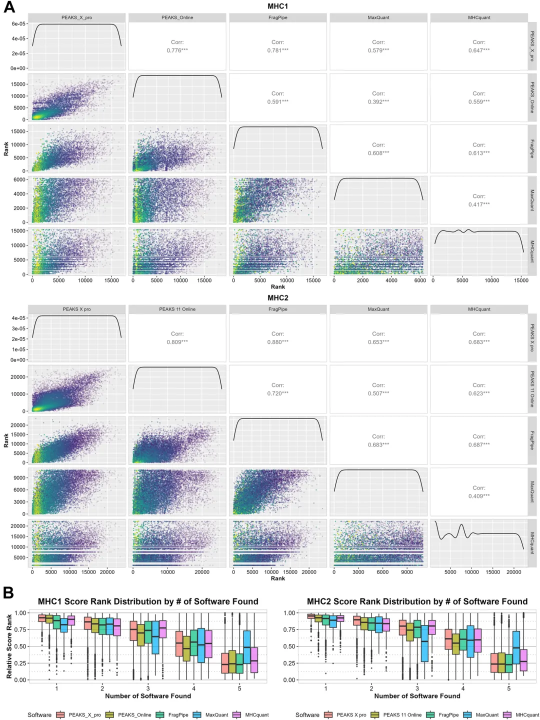
Figure 3 置信度得分相关性
为了评估不同软件的 FDR 质控效果,作者额外加入拟南芥的数据库作为诱饵库,与原有 Target-decoy 方法对比,Figure 4 结果显示,在肽段水平的 1% FDR 阈值下,拟南芥诱饵库方法在所有软件中均获得了更高的鉴定数量,而且这种差距随着 FDR 的增加而扩大。这表明对于所有软件来讲,常规的 Target-Decoy 策略即可以实现无偏估计。
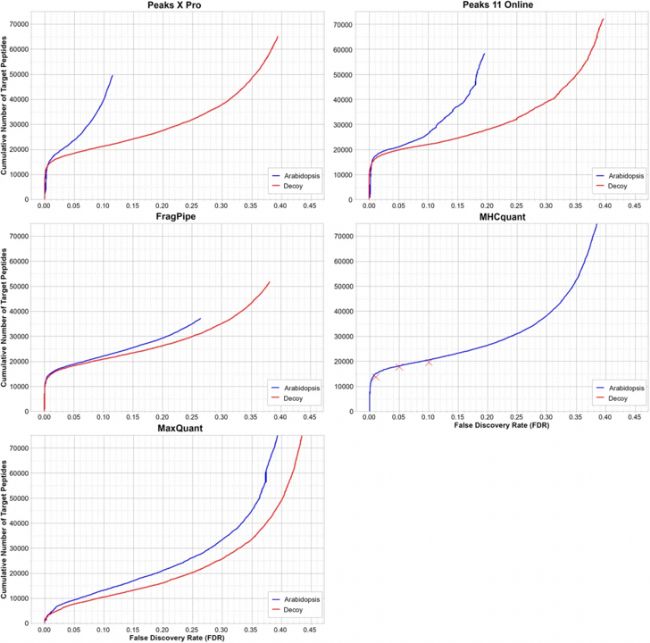
Figure 4 拟南芥诱饵库FDR质控对比
从前面的 FDR 测试可以看出,参考数据库是蛋白质组学搜索中的关键参数,它通过影响 FDR 过滤和评分来影响肽段鉴定的一致性和准确性。
因此,作者进一步通过系统地测试不同大小的数据库来探究这一问题,以揭示软件如何适应不断变化的搜索空间,从而揭示出使用单一固定数据库时不易察觉的优势和局限性。通过 Figure 5 的统计结果可以看出,在常规 Uniprot 数据库的基础上,加上转录组和DNTA序列后,在 1% FDR 阈值下,基本上所有软件的鉴定结果反而降低了,因此,在分析免疫肽组数据时,并非数据库越大越好。标准 UniProt 库分析下,DeepNovo Peptidome workflow的工作性能依然是最佳的。另外,作者通过识别单氨基酸位点突变(SAV)及其对应参考序列的成对组合来评估软件区分SAV的能力。所有工具均成功检测到此类成对组合,其中 PEAKS® 软件报告的此类变异的数量及比例均最高,尤其是在包含 DNTA 时。
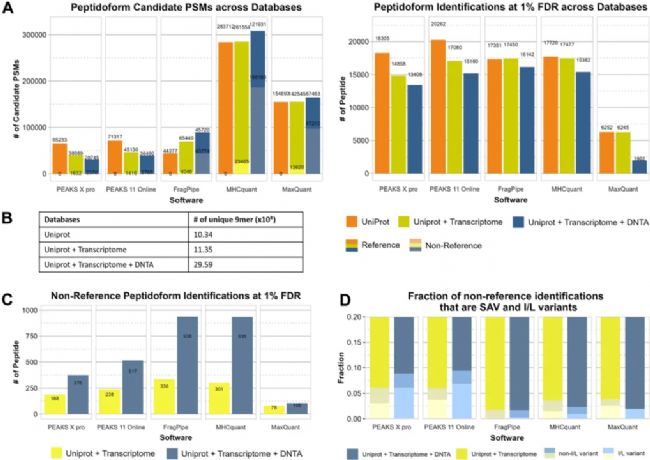
Figure 5 不同数据库搜索结果对比
小结
本篇MCP基准研究为免疫肽组学质谱数据分析提供了非常全面的参考信息,综合多个指标来看,DeepNovo Peptidome workflow 是当前免疫肽组学的分析的最优选择,为肿瘤新抗原、疫苗靶点、免疫表位研究提供最灵敏、最可靠、最稳定的生信解决方案。
文献信息
标题:Benchmarking Software for DDA PASEF Immunopeptidomics
期刊:Mol Cell Proteomics, 2026, 25(4)
DOI:10.1016/j.mcpro.2025.101492
感兴趣的老师还可以关注我们即将发布的全新peptidome workflow,其中DDA peptidome 工作流新增PEAKS openPTM的开放搜索算法,DIA Peptidome支持突变分析,并新增内标肽导入和分析功能。
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
- 免疫肽由胞内多种蛋白酶剪切产生,无固定酶切特异性,使得搜索空间呈指数级扩大;
- 肽段长度分布宽 (MHC Ⅰ 8–13 AA、MHC Ⅱ 13–25 AA)、翻译后修饰多样;
- 抗原肽丰度差异极大,且常伴随同分异构、单氨基酸变异(SAV)等复杂情况;
- 现有的大部分搜库软件多为常规蛋白质组设计,缺乏针对免疫肽数据的系统基准验证。
软件选择不当会直接导致高置信抗原漏检、假阳性偏高、变异肽识别不准、实验数据无法充分挖掘等问题的发生。
针对这一痛点,德国癌症研究中心免疫肽组学研究平台的Stefan Tenzer教授携团队在 MCP 发表重磅基准测试结果。以JY细胞系MHC Ⅰ/Ⅱ免疫肽为标准样品,基于 Thunder DDA PASEF 采集质谱数据,对当前五大主流搜库软件开展深度对比。
结果表明 PEAKS® 的 DeepNovo Peptidome workflow 对免疫肽组学研究具备强大的综合分析能力,为全球免疫肽组学研究者提供了最直观的软件选择与流程优化依据。
本次基准测试对比了 PEAKS® Online 11, PEAKS Studio Xpro, FragPipe, MaxQuant 和 MHCquant 五个搜库软件,均设置1% PSM FDR。
Figure 1A 的整体统计结果显示,使用PEAKS® Online 11内置的 DeepNovo Peptidome workflow 鉴定到的总数最高,MHC Ⅰ/Ⅱ肽段较 PEAKS Studio X Pro中的传统 DB Search 和 FragPipe 高出 6%–11%。FragPipe 紧随其后在开源软件中表现最佳,MHCquant 表现稳健,略低于 FragPipe,MaxQuant 鉴定量最低,较 MHCquant 少 50%–60%。Figure 1 交集分析显示,除 MaxQuant 外,其他软件间的重合度约 58%-60%,说明主流工具对高置信肽判断一致,整体无极端偏好偏差。Figure 1C-D 表明所有软件对母离子电荷及肽段长度分布均高度一致,无系统性偏差。
(新版 PEAKS ®Online 与 PEAKS® Studio 已实现算法统一,均支持DeepNovo Peptidome的肽组学专属工作流,用户可在任一平台获得同等高性能表现。)
Figure 1 不同软件的MHC Ⅰ&Ⅱ综合鉴定结果对比
随后,作者对所有软件鉴定到的多肽通过 netMHCpan 和 netMHCIIpan 分别对MHC-Ⅰ和MHC-Ⅱ型多肽进行结合强度预测,Figure 2 结果显示,PEAKS ®DeepNovo Peptidome workflow鉴定到的总结合肽数量最多,覆盖更广;MHCquant 结合肽本身比例更高,更偏向于高特异性结合肽段。各软件氨基酸组成高度保守,且样本重现性基本稳定,仅 MaxQuant 有少量显著偏差。
Figure 2 免疫肽结合力和重现性对比
同时,作者对比了 5 款软件共同鉴定到肽段的置信度得分 Spearman 相关系数,结果显示越多软件共有的多肽,相关性排名越靠前。
Figure 3 置信度得分相关性
为了评估不同软件的 FDR 质控效果,作者额外加入拟南芥的数据库作为诱饵库,与原有 Target-decoy 方法对比,Figure 4 结果显示,在肽段水平的 1% FDR 阈值下,拟南芥诱饵库方法在所有软件中均获得了更高的鉴定数量,而且这种差距随着 FDR 的增加而扩大。这表明对于所有软件来讲,常规的 Target-Decoy 策略即可以实现无偏估计。
Figure 4 拟南芥诱饵库FDR质控对比
从前面的 FDR 测试可以看出,参考数据库是蛋白质组学搜索中的关键参数,它通过影响 FDR 过滤和评分来影响肽段鉴定的一致性和准确性。
因此,作者进一步通过系统地测试不同大小的数据库来探究这一问题,以揭示软件如何适应不断变化的搜索空间,从而揭示出使用单一固定数据库时不易察觉的优势和局限性。通过 Figure 5 的统计结果可以看出,在常规 Uniprot 数据库的基础上,加上转录组和DNTA序列后,在 1% FDR 阈值下,基本上所有软件的鉴定结果反而降低了,因此,在分析免疫肽组数据时,并非数据库越大越好。标准 UniProt 库分析下,DeepNovo Peptidome workflow的工作性能依然是最佳的。另外,作者通过识别单氨基酸位点突变(SAV)及其对应参考序列的成对组合来评估软件区分SAV的能力。所有工具均成功检测到此类成对组合,其中 PEAKS® 软件报告的此类变异的数量及比例均最高,尤其是在包含 DNTA 时。
Figure 5 不同数据库搜索结果对比
小结
本篇MCP基准研究为免疫肽组学质谱数据分析提供了非常全面的参考信息,综合多个指标来看,DeepNovo Peptidome workflow 是当前免疫肽组学的分析的最优选择,为肿瘤新抗原、疫苗靶点、免疫表位研究提供最灵敏、最可靠、最稳定的生信解决方案。
文献信息
标题:Benchmarking Software for DDA PASEF Immunopeptidomics
期刊:Mol Cell Proteomics, 2026, 25(4)
DOI:10.1016/j.mcpro.2025.101492
感兴趣的老师还可以关注我们即将发布的全新peptidome workflow,其中DDA peptidome 工作流新增PEAKS openPTM的开放搜索算法,DIA Peptidome支持突变分析,并新增内标肽导入和分析功能。
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,ProteoformXTM,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
相关文章
更多 >